Cleanrooms are contamination-free environments where high-tech manufacturing and assembly take place. Cleanrooms range from very small chambers, called microenvironments, to large-scale rooms, called ballrooms. Cleanroom technology is used in a wide range of industries including semiconductor assembly, biotechnology, pharmaceutical, aerospace, food, medical devices and hospitals. There are a number of important aspects to consider while determining which cleanroom type fits the needed application. This includes the cleanliness class, fabrication type, and special features such as ESD control, pass throughs, and a gowning area. Cleanliness class is a standard determined by the contamination control industry. They currently use a government specification known as Federal Standard 209D to provide a qualified and standardized method for measuring how clean the air is in a cleanroom. Six classes have been established to designate cleanroom cleanliness.
range from very small chambers, called microenvironments, to large-scale rooms, called ballrooms. Cleanroom technology is used in a wide range of industries including semiconductor assembly, biotechnology, pharmaceutical, aerospace, food, medical devices and hospitals. There are a number of important aspects to consider while determining which cleanroom type fits the needed application. This includes the cleanliness class, fabrication type, and special features such as ESD control, pass throughs, and a gowning area. Cleanliness class is a standard determined by the contamination control industry. They currently use a government specification known as Federal Standard 209D to provide a qualified and standardized method for measuring how clean the air is in a cleanroom. Six classes have been established to designate cleanroom cleanliness.
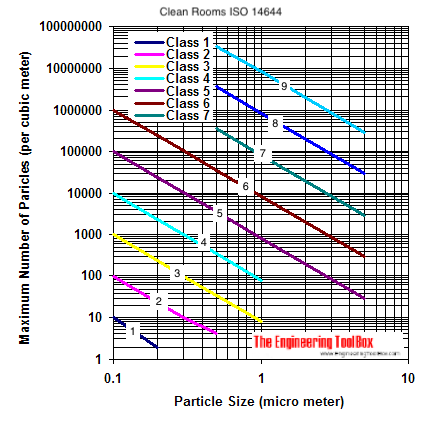
The class number refers to the maximum number of particles bigger than one-half of a micron that would be allowed in one cubic foot of cleanroom air. A Class 100 cleanroom, for example, would not contain more than 100 particles bigger than half a micron in a cubic foot of air. The six classes are Class 1 (ISO 3), Class 10 (ISO4), Class 100 (ISO 5), Class 1,000 (ISO 6), Class 10,000 (ISO 7), and Class 100,000 (ISO 8)
There are five main fabrication styles for cleanrooms (although custom styles are available). They are conventional, modular hardwall, modular softwall, mini environment, and micro environment. Conventional construction is the most common type, and these are generally permanent structures.
Modular cleanrooms are constructed on site from pre-cut and assembled components, such as walls, ceiling grid struts and other components. Hardwall cleanrooms provide the rigidity and durability of a freestanding room. Their walls of the cleanroom are of a solid material, rather than fabric. The walls of modular softwall cleanrooms are constructed from fabric, either of free-hanging strips or stretched tightly over a frame.
Mini environments are localized clean environments are used in semiconductor manufacturing applications. They are created around a specific tool, or only within a tool, to protect the semiconductor wafer from atmospheric exposure. Wafers are moved from one mini environment equipped tool to another in sealed containers and only exposed to the atmosphere inside of the clean mini environment. Micro environments are similar, but they are smaller, are used to protect the wafer itself, or a part thereof, instead of encapsulating the manufacturing tool.
Some cleanrooms are available with Electrostatic Discharge (ESD) control. A cleanroom with ESD control is able to measure and contain electrical discharge thereby avoiding rapid, spontaneous and usually uncontrolled transfer of an electrical charge between two conductors induced by a strong electrostatic field.