Introduction
The microbiological quality of drugs and biologics
is necessary for their efficacy and patient safety, because microbial
contamination of drugs causes immediate adverse effects on patient
health in terms of morbidity and mortality,
1-3 as well as
long-term adverse effects, such as cancer, autoimmune, and other
diseases. Additionally, microbes can alter the chemistry and
pharmacology of drugs, with a potential adverse impact on their
effectiveness due to the breakdown of the active ingredients as well as
on their safety due to the toxicity of potential degradant products.
Therefore, control of microbes in drugs is essential, either by assuring
absence of microbes in sterile drugs that are administered parenterally
and applied to sensitive tissues or by controlling microbial bioburden
to appropriate levels for nonsterile drugs that are administered to
regions rich in microbial flora with physical or immunological barriers
to infections.
Table 1 lists major differences between sterile
and non-sterile drugs. For sterile drugs, microbes are essentially
eliminated by terminal sterilization (heat or irradiation of final
containers) or by employing an aseptic manufacturing process where
terminal sterilization is not possible, specifically for most biologics.
Assurance of the absence of bacterial, yeast, and fungal contaminants
is provided by the sterility test for sterile drugs.
4 For
non-sterile drugs, bioburden due to aerobic bacteria, yeast, and fungi
and absence from objectionable microorganisms, as required, is
controlled to appropriate levels based on product attributes, route of
administration (oral, intranasal, topical, anal, vaginal, etc) and
target patient population (neonates, infants, elderly,
immunocompromised, healthy population, etc). Non-sterile drugs are
tested for total aerobic bacteria, yeast, and fungi by the bioburden or
microbial limit test
5-7 and for the absence of objectionable organisms,
6-11 as required (
Table 1).
Table 1. Major Differences between Sterile and Non-Sterile Drugs and Biologics

Limitations of Microbiological Testing
Microbiological
testing plays a significant role in assuring the appropriate quality of
drugs. However, the paradigm of final product testing, particularly for
microbiological quality, is shifting, because testing alone does not
provide complete or absolute assurance for control or absence from
microbes (eg, bacteria, fungi, mycoplasma, and viruses). Additionally,
the reliability of microbiological testing depends upon the selection of
appropriate methods that are “Suitable for Intended Purpose” and an
adequate number of samples taken at appropriate stages of manufacture.
12
For example, to provide an absolute assurance for the absence of
microbes in a product, the whole product will be required to be tested
for sterility. After the test, there will be no product for actual
therapeutic use.
Building Microbiological Quality into Drugs
Microbiological
quality needs to be built into the drugs by understanding the sources
of contamination, environmental conditions, and product attributes that
support growth of microbes. Microbiological quality for sterile drugs is
assured by employing a robust environmental monitoring (EM) program,
appropriate microbiological testing at various stages or intermediate
products during manufacture, including the final drug product (DP) and
using validated manufacturing processes (eg, aseptic manufacturing
processes, container closure studies, media fill studies, etc). During
routine manufacture of sterile drugs employing aseptic manufacturing
processes, EM is an essential and critical component to demonstrate the
state of control of the facility, providing information on the microbial
quality of manufacturing and testing environments. This is an important
element for sterility assurance of sterile drugs. There are a number of
guidance documents and regulations on the EM aspects of manufacture of
sterile drugs.
13-15 Microbiological quality of nonsterile
drugs is important, too, and can be assured through selection of
appropriate controls through a risk analysis process. Many sterile drugs
have certain components or intermediate products that are classified as
non-sterile and are manufactured like non-sterile drugs. Therefore,
understanding the risk of introduction of microbes and their products
(such as toxins and proteases) during manufacture of non-sterile drugs,
and intended use of the product in a target population (such as use of
vaccines in healthy individuals) are important considerations in
choosing a manufacturing process—sterile or non-sterile. There are
expectations and a need to control and monitor the environment for
manufacture of non-sterile drugs, intermediate products, or components.
6,8,16,17
However, there is not much guidance or clarity on regulatory
expectations on the EM program for non-sterile drugs. Recently the
United States Pharmacopoeia (USP) drafted guidelines to monitor the
environment for manufacture of such drugs.
18 These guidelines describe a risk-based approach to control microbes for manufacture of non-sterile drugs.
In
this article, the role of EM and microbiological testing in eliminating
or controlling primarily bacterial, yeast, and fungal contaminants
during manufacture of drugs and biologics—specifically vaccines—is
discussed. Control and testing for adventitious viruses, mycoplasma,
residual live viruses or bacteria, and other aspects of microbiological
testing critical in the safety of biologics, are not covered in this
article. Recently, there have been significant concerns and discussions
about the sterility assurance of drugs formulated by compounding
pharmacies and microbial control during such operations due to a number
of adverse events, including deaths from use of fungal contaminated
methylprednisolone injections.
3 This article does not cover
microbiological quality of drugs made by compounding pharmacies. The USP
has several chapters on controlling microbiological quality of such
drugs.
19-22
Challenges in Assuring Microbiological Quality for Biologics
As
discussed above, microbiological quality of drugs and biologics is
critical for their safety and effectiveness. But biologics, particularly
vaccines, pose unique and complex challenges in achieving
microbiological quality (
Table 2). Biologics, as per their definition, are made from starting materials that are biological in nature and support microbial
growth during the manufacturing process, creating challenges in
maintaining sterility or purity of the desired organism. Many biologics
are made in embryonated eggs, animals, and cells of avian, mammalian, or
insect origin, collectively referred to as the substrate, which may
contain inherent adventitious agents and support the growth of microbial
contaminants. Starting materials, such as seed viruses or bacterial
seed stock cultures, may consists of pathogenic or attenuated bacteria
or viruses posing a risk to the operators, environment, and the final
product due to presence of residual live bacteria or viruses and active
toxins. Further, several raw materials, such as growth media, fetal
bovine serum, trypsin, etc, used during manufacture of biologics are of
animal origin. All of these components (substrate, seed stocks, raw
materials, etc) pose substantial risks of inherent contaminants and
adventitious agents, which may grow during manufacture of the product or
grow in the human body after administration of the product. Therefore,
all these components require documented history of their origin or
isolation and passage history with complete traceability (ie, exposure
to various reagents during isolation and propagation). Extensive testing
for inherent and adventitious agents, including viruses, mycoplasma,
bacteria, yeast, and fungi, and risk analysis for bovine spongiform
encephalopathy and transmissible spongiform encephalopathies, are
performed on seed stocks, cell banks, batches of media components, etc,
at various stages (ie, master and working cell banks or seed stocks,
harvests, or other intermediate stages during manufacture). Aseptic
manufacturing process seems essential for manufacture of biologics due
to the risks discussed above and also due to the fact that biological
products being proteins, polysaccharides, carbohydrates, lipids, etc,
and growth media (used during manufacture or as a residual component in
intermediate components or final product) support microbial growth.
Table 2. Challenges in Achieving Mmicrobiological Quality for Vaccines and Need for Aseptic Processes for Manufacturing Vaccines

Regulation of Biologics and their Microbiological Quality
The
challenges in assuring microbiological quality of biologics have been
recognized by regulatory agencies around the world for decades, and
additional or separate requirements have been in place to regulate
biologics.
23-26 The US Parts 600 to 680 of 21 Code of Federal Regulations (21 CFR 600–680) describe the regulation of biologics.
23
Recognizing the microbial contamination risk during manufacture of
biologics, 21 CFR 610.12 specifically required sterility testing on
final bulk or Drug Substance (DS) of biologics.
27 In
practice, biologics, particularly vaccines that are given to millions of
healthy babies and infants, sterility tests have been performed at a
number of intermediate products, including final bulk, to achieve
maximum sterility assurance for vaccine products. In 2012, the sterility
test described for biological products in 21 CFR 610.12 was amended to
exclude testing at final bulk stage.
28 This change could be a
significant risk for contamination of vaccines given to healthy
individuals and may subsequently lead to adverse reactions in
recipients. Until 2012, all biological products had to be sterile from
final bulk stage or earlier, usually manufactured aseptically following
processes for making sterile drugs. With the amended sterility
requirement, the final bulk does not need to be manufactured aseptically
and can be manufactured following processes used for making non-sterile
drugs. European Pharmacopeia (Ph. Eur.) chapter 7.6 allows replacement
of the sterility test for intermediate products with a bioburden test
having low limit specifications, with the conditions that intermediate
product can be filter-sterilized and the intermediate product does not
support microbial growth during storage.
29 This requirement
can be applied on a case by case basis, based on the risk-benefit ratio
and after meeting conditions discussed in Ph. Eur. However, replacing
the sterility test for vaccines with a bioburden test at intermediate
product and final bulk stages, leading to selection of non-sterile
manufacturing processes for vaccines, is not scientifically and
technically sound (discussed later).
Table 2 summarizes major
reasons for employing aseptic processes for manufacturing vaccines. Not
employing aseptic manufacturing processes will lead to lack of sterility
assurance achieved through EM and aseptic process validation that are
not required for the manufacture of non-sterile drugs or components.
Further, a bioburden test does not require testing for anaerobic
bacteria,
5,6 such as
Clostridium tetani, Clostridium botulini,
etc, which produce lethal toxins. There is a potential risk of
contamination with such toxins of products made by non-sterile
manufacturing process and not tested for absence of anaerobic bacteria.
General Principles to Control Microbes during Manufacture of Sterile and Non-Sterile Drugs
Building,
monitoring, and maintaining cleanroom environment is expensive, and it
may not be required or desirable for non-sterile drugs if there is no
value for the patient. A careful risk analysis is required to make a
decision considering the unique challenges posed by the manufacture of
biologics and the use of vaccines in a healthy population as discussed
above (
Table 2). In contrast to biologics, drugs are usually
chemical salts or compounds, often in dry powder form, and do not
support growth of microbes during storage, even at room temperature.
Therefore, low level of bioburden is usually acceptable, particularly
when these drugs are meant for topical, oral, or intranasal use. There
is not much risk in using non-sterile processes to manufacture
intermediate products or active pharmaceutical ingredients (API) for sterile drugs because these APIs
are usually in powder form, and do not support growth of microbes.
There are certain general principles to control microbes for manufacture
of both sterile and non-sterile drugs.
- Microbial growth in excipients, APIs, components, and DS should be monitored and controlled to avoid unacceptable levels.
- Microbial
growth is not only a risk for microbial toxins or other toxic
components produced during growth, but could also damage the chemical
and pharmacological properties of drugs.
- In particular, microbial proteases could break down proteins in biological products.
- Manufacturing,
testing, and storage facilities should not have any microbial growth,
which can be a source of contamination for the raw materials,
intermediate products, DS, and DP.
- Manufacturing and testing
facilities should have controlled access with procedures in place to
control or prevent entry of microbes in the facility.
- Lower
bioburden levels in DP, components, and raw materials than those
required in compendia and product not supporting microbial growth at the
recommended storage conditions will control the risk of microbial
toxins, and ensure the stability of drugs from microbial degradation.
Aseptic Manufacturing Process and Environmental Monitoring
Table 3. Essential Elements of Aseptic Manufacturing Process

Table 3
summarizes essential elements of aseptic process for the manufacture of
sterile drugs. A robust EM program is an essential and critical
component to demonstrate the state of control of the facility and the
environment required for an aseptic manufacturing process. However, EM
is not a direct measure of batch sterility due to inherent variability
of methods used to monitor the environment and also due to a lack of a
correlation between EM levels and batch sterility.
30
Nonetheless, EM provides valuable information about the status of
cleanrooms, whether meeting required specifications with regard to
particles and viable organisms, the performance of HVAC system, use of
acceptable personnel techniques, gowning practices, status of the
equipment, and cleaning operations.
30 A number of regulatory guidance documents
13,14,25 and a recent publication
30provide
valuable information about the aseptic manufacturing process and
requirements for an eff ective and robust EM program. This article is
not intended to go into details of all aspects related to aseptic
manufacturing. Instead, this article highlights some important aspects
that need discussion, particularly aspects important in the manufacture
of biologics.
Cleanrooms
Selection grade or class of
cleanroom for each stage of manufacture of biologics is complex and one
of the most misunderstood areas in implementing cGMP regulations. A
thorough risk assessment approach is an important cGMP tool for an eff
ective EM program.
Table 4 lists essential components of EM. A
basic element of an EM program is the classification of cleanrooms.
Currently, there are 3 major systems for the classification of
cleanrooms used in the pharmaceutical industry based on the number of
air particles >0.5 μ in a cubic foot of air.
13-15,25,31
For example, the critical area of aseptic manufacture, Class 100, should
not have more than 100 particles of ≥0.5 μm in one cubic foot of air.
As per International Standard Organization (ISO) this area is classified
as ISO 5,
31 which is equivalent to Grade A of European
Union’s (EU) GMP guidelines, classified on the basis of metric system
(not more than 3520 particles of ≥0.5 μm in one cubic meter of air) and
EU grading of cleanrooms is based on counts during operations and at
rest.
14 Classification of cleanroom is a universal standard,
not only for the pharmaceutical industry, but also for other industries
(such as electronics) and has been described elsewhere.
13-15,25,31
One of the major differences in various regulatory guidance and
requirements is the classification of the supporting area for the
critical Class 100 area. The FDA guidance document suggests Class 100 in
Class 10,000 (ISO 7 or Higher),
13 whereas EU GMP requirements and WHO guidelines recommend class 100 in class 1000 or ISO 6.
14,25
Class 1000 and class 10,000 areas have significant different
specifications for EM parameters, particularly viable organisms. In
older vaccine manufacturing facilities, it is sometimes difficult to
meet class 1000 specifications for upstream manufacturing processes when
there are supporting data on the aseptic process from the purity of a
culture during fermentation. The intermediate product is immediately
sterile-filtered after confirming purity or bacteriostatic
preservatives, inactivating agents, such as formaldehyde are added to
detoxify toxins, to inactivate bacteria or viruses or to avoid
contamination. Such risk analysis and the impact on the quality of the
product will be useful to justify a change in supporting area from Class
1000 to Class 10,000. Use of Class 10,000 supporting area for Class 100
critical area with a risk analysis, as discussed, will be a stringent
control than the current regulations of not requiring sterility at the
final bulk, leading to classifying the manufacturing process as
non-sterile.
Table 4. Major Components of Environmental Monitoring
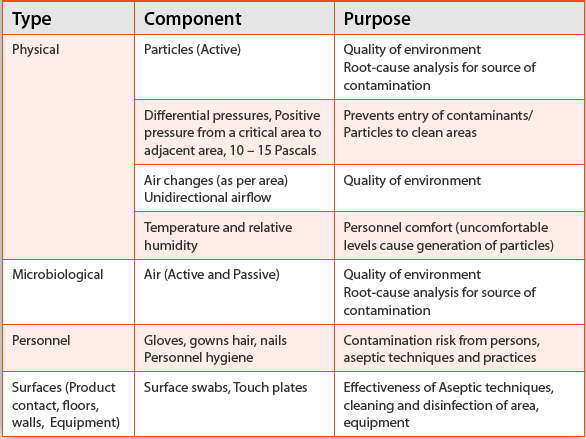
Environmental Monitoring
Table 4
lists major components of an EM program. It is important to understand
these components, which will help in the selection of appropriate
methods to implement an effective and robust EM program. Evaluating the
quality of air, surfaces, personnel, etc, in a cleanroom environment
should start with a well-defined written program employing
scientifically sound methods of sampling, testing, data analysis, etc,
with an independent oversight by the quality assurance department.
Sampling locations and adequate sampling are critical components of an
effective EM and should be specified in the written program or standard
operating procedures.
30 For example, air and surface samples need to be taken at locations with significant activity or product exposure.
Air Monitoring
Air
monitoring for total particles is usually done for 0.5- to 5- and
>5-μm particles as the cleanrooms are classified based on these
counts. Currently, on-line air monitoring systems using remote probes
are available to count particles on a continuous basis, both statically
and during operations (dynamically). Manual air samplers with
well-defined and documented sampling locations, volume of air to be
sampled, and sampling frequency may also be used. Sampling locations and
placement of probes should be carefully evaluated to collect
information that provides status on the quality of the environment
during operations. Viable particles (microbes) can be monitored either
actively using air samplers or passively by settle plates.
30
Historically, microbes have been monitored for aerobic bacteria, yeast,
and fungi. Several firms have been using anaerobic incubations of media
plates to isolate anaerobic bacteria from cleanroom environments.
Personnel Monitoring
Personnel
are the largest risk factor in aseptic manufacturing processes. During
each session, gloves and gowns are periodically sampled and monitored
for aerobic bacteria, yeast, and fungi with a need to monitor for
anaerobic bacteria—particularly
Propionibacterium acnes, a
facultative anaerobe, which is part of the skin normal flora and has
been isolated from manufacturing environments. Personnel health
monitoring and medical examination are required for those working in
aseptic manufacturing processes. Normal flora from these persons,
particularly from nails, hands, hair, etc, may be useful during
investigations to find out the source of contamination.
Personnel Training
All
operators should be trained and qualified on various procedures,
including gowning, with a good understanding of the procedures, their
importance in aseptic manufacturing operations, and the impact or risk
to quality for not following these procedures. Training on working in a
cleanroom should focus on minimizing the generation of particles and
disruption of air flow. Examples of personnel training can include
aseptic technique, cleanroom behavior, microbiology, hygiene, patient
safety hazards due to non-sterile drugs, and specific written procedures
on manufacturing operations. For general techniques and operations in
cleanrooms, emphasis should be placed on contacting sterile materials
with sterile instruments only, with no direct contact of sterile
products, containers, closures, or critical surfaces with gown or
gloves. In a critical cleanroom area (Class 100), personnel movements
should be slow and deliberate in order not to disrupt unidirectional
airflow and to avoid turbulence. The entire body should be kept out of
the path of unidirectional airflow with a proper gown control.
Surfaces
Samples
from surfaces by touch plates or surface swabs are monitored for viable
microbes to evaluate the effectiveness of operations, cleaning, and
disinfection procedures. Critical surfaces coming in contact with a
sterile product should remain sterile throughout an operation.
Analysis of Data and Follow-ups
All
EM data should be trended and tracked in real time with the
establishment of appropriate alert and action levels based on regulatory
guidelines, requirements, and risk-benefit analyses of the product.
Averaging the results of EM samples can mask unacceptable conditions.
Investigations
for excursions and changes in microbial flora should be thorough with
an emphasis on determining the root cause. EM should promptly identify
potential root cause of contamination, allowing for implementation of
corrections before product contamination occurs.
13,30 EM is
important to monitor the microbiological quality of critical areas to
determine if aseptic conditions are maintained during manufacturing
operations.
Selection of Sterile or Non-sterile Manufacturing Process
Sterile
drug manufacturing process requires a sterility test at the end to
demonstrate the absence of any viable bacterial, yeast, and fungal
contaminant. Non-sterile drug manufacturing process requires a bioburden
test to provide the number of viable aerobic bacterial, yeast, and
fungal organisms that should be lower than the specifications, and
absence of objectionable organisms. With the elimination of sterility
test at final bulk or DS or replacement of sterility test with the
bioburden test, the manufacturing process for that intermediate product,
final bulk or DS will be a non-sterile process. This is an important
change in the manufacture of biologics, particularly vaccines (
Table 2).
Lack of much guidance on microbiological controls, no requirements for
classification of cleanrooms, and no requirements for EM in manufacture
of non-sterile drugs will result in a significant risk to the
microbiological quality of vaccines. Can vaccines be manufactured by
non-sterile manufacturing process until the final bulk or DS, then
filter-sterilized for filling? Answering yes to this question seems
scary. This is an example where we need to go ‘back to basics’ and
return to the science of applied or pharmaceutical microbiology, as
emphasized by Lolas in a recent commentary.
32 Based on this
author’s experience and knowledge in the manufacture and regulation of
vaccines, these products cannot be manufactured like nonsterile drugs
and then sterile-filtered at the final bulk before filling. From a
historical, technical, and scientific perspective as summarized in
Table 2,
vaccines have been made under aseptic conditions. Sterile manufacturing
process for vaccines will also be supported from a business perspective
due to a high risk of microbial contamination during manufacture (
Table 2).
Microbial contamination will have significant impact on the yield and
quality of the final product, leading to rejection of a number of DP
lots. Immunization with vaccines has been one of the most successful and
cost-effective public health interventions in controlling infectious
diseases.
33 Changing the manufacturing of vaccines to
non-sterile processes has the potential risk of a major public health
disaster that will shatter the confidence of the public in safety of
vaccines.
Sterility Test and Bioburden Test
As discussed above, a sterility test is required for sterile drugs4 and a bioburden test is required for non-sterile drugs.
5,6
Both tests have the limitations of microbiological methods and do not
provide absolute results or a complete assurance on the absence of
viable organisms. There are some other differences in these tests, which
are important to understand, which will provide further rationale that
vaccines should not be manufactured by non-sterile processes.
For
the sterility test of intermediate products and final bulk, a 10-ml
sample is tested in each of 2 media. The amount of sample for a
bioburden test depends upon the specification to provide the assurance
for that specification. There has been a significant misunderstanding
and confusion in setting specifications for a bioburden test and
expressing the results. A specification of <1 0.1-ml="" 0.1="" 0="" 1-="" 100="" 10="" 1="" a="" absence="" almost="" an="" and="" any="" are="" as="" assurance="" at="" bacterial="" be="" because="" been="" bioburden="" bulk="" but="" by="" cause="" cfu="" cfus="" colony="" comparing="" considered="" counts="" determine="" discussed="" do="" does="" drugs="" emphasizes="" end="" established="" even="" example="" expressed="" expressing="" expression="" filtration="" final="" for="" forming="" from="" further="" has="" importance="" in="" inaccurate="" intermediate="" interpreted="" is="" lesser="" level="" liquid.="" load="" low="" manufacture="" materials="" may="" microbes.="" microbes="" microbiologists="" microbiology="" million="" misunderstanding="" ml.="" ml="" must="" no="" non-microbiologists="" non-sterile="" not="" of="" often="" on="" or="" particularly="" per="" perspective.="" product="" products="" provide="" raw="" recently.="" regulatory="" required="" respectively="" results="" sample="" scientifically="" specification="" statistically="" sterile.="" sterile="" sterility="" such="" sufficient.="" suitable="" sup="" test="" tested.="" the="" there="" these="" this="" times="" to="" turbidity="" units="" up="" very="" visible="" when="" will="" with="">32
The Ph. Eur. suggests replacing sterility test with a bioburden test
for intermediate products during manufacture of vaccines with a low
bioburden specification and when product does not support growth of
microbes.
29A low bioburden specification should be <0 .1="" 10-ml="" 10="" a="" absence="" as="" assurance="" be="" bioburden="" bulk.="" cfu="" despite="" fact="" final="" for="" in="" intermediate="" level="" low="" manufacturing="" ml="" of="" on="" or="" p="" per="" performed="" process="" product="" provide="" require="" sample="" should="" similar="" specification.="" specification="" sterile="" sterility="" still="" such="" test="" testing="" that="" the="" this="" to="" which="" will="" with="">Finally, lack of testing for anaerobic bacteria in the
bioburden test is a major limitation and could be a potential risk of
contamination of the product with deadly microbial toxins as discussed
earlier.
Summary
Environmental monitoring and
microbiological testing play a critical role in ensuring the safety of
patients and the efficacy of drugs and biologics by preventing their
contamination with microbes. Microbiological testing alone does not
provide complete or absolute assurance of absence of microbial
contamination. However, such testing combined with a robust environmental monitoring
program and the use of validated manufacturing processes provides a
high degree of assurance of the microbial safety of drugs. To build
microbiological quality in drugs and biologics, it is important to
understand the ways to prevent contamination and risks of microbial
growth in intermediate products, components, active pharmaceutical
ingredients, final bulk or drug substance, and final product.
Manufacturing processes (sterile or non-sterile) should be based on
factors such as risk analysis, target population for the drug, and the
route of injection.
Rajesh K. Gupta, PhD, is Principal Consultant, Biologics Quality & Regulatory Consultants, LLC, North Potomac, MD 20878, USA
References
- Matthews
BR. The Devonport incident, the Clothier Report, and related matters –
30 years on. PDA J Pharm Sci Technol. 2002;56(3):137-49.
- Arie S. Contaminated drugs are held responsible for 120 deaths in Pakistan. BMJ 2012;344:e951, doi: 10.1136/bmj.e951.
- Smith
RM, Schaefer MK, Kainer MA, et al. Fungal Infections Associated with
Contaminated Methylprednisolone Injections. N Engl J Med. 2013;
369:1598-1609. doi: 10.1056/ NEJMoa1213978.
- USP <71> Sterility Tests. Current Version, The United States Pharmacopeial Convention, Rockville, MD.
- USP
<61> Microbial Examination of Nonsterile Products: Microbial
Enumeration Tests. Current Version, The United States Pharmacopeial
Convention, Rockville, MD.
- USP <1111> Microbiological
examination of nonsterile products: Acceptance criteria for
pharmaceutical preparations and substances for pharmaceutical use.
Current Version, The United States Pharmacopeial Convention, Rockville,
MD.
- European Pharmacopeia, Chapter 5.1.4, Microbiological
Quality of non-sterile pharmaceutical preparations and substances for
pharmaceutical use. Current version, Quality of Medicines and
Healthcare: Strasbourg, France.
- USP <62> Microbiological
Examination of Nonsterile Products: Tests for Specified Microorganisms.
Current Version, The United States Pharmacopeial Convention, Rockville,
MD.
- Code of Federal Regulations, Title 21 Food and Drugs, Part
211.84(d)(6). Testing and approval or rejection of components, drug
product containers, and closures. Washington, DC: US Government Printing
Office; April 2014. Available at: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm?CFRPart=211&showFR=1 Accessed September 27, 2014.
- Code
of Federal Regulations, Title 21 Food and Drugs, Part 211.113(a).
Control of microbiological contamination. Washington, DC: US Government
Printing Office; April 2014. Available at: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm?CFRPart=211&showFR=1 Accessed September 27, 2014.
- Code
of Federal Regulations, Title 21 Food and Drugs, Part 211.165(b).
Testing and release for distribution. Washington, DC: US Government
Printing Office; April 2014. Available at: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm?CFRPart=211&showFR=1 Accessed September 27, 2014.
- Gupta RK. Validation of Microbiological Methods – Expectations for Regulatory Compliance. BioPharm Asia. 2014 (in press).
- FDA
Guidance for Industry-Sterile Drug Products Produced by Aseptic
Processing - Current Good Manufacturing Process, 2004. Available at http://www.fda.gov/downloads/Drugs/ GuidanceComplianceRegulatoryInformation/Guidances/UCM070342.pdf Accessed September 27, 2014.
- EU
Guidelines to Good Manufacturing Practice, Medicinal Products for Human
and Veterinary Use, Volume 4, Annex 1, Manufacture of Sterile Medicinal
Products, 2008, Available at http:// ec.europa.eu/health/files/eudralex/vol-4/2008_11_25_gmp-an1_en.pdf. Accessed September 27, 2014.
- PIC/S,
Pharmaceutical Inspection Convention Guide to Good Manufacturing
Practice for Medicinal Products Annex 1, Manufacture of Sterile
Medicinal Products, 2014.
- Code of Federal Regulations, Title 21
Food and Drugs, Part 211.80(b). General Requirements. Washington, DC: US
Government Printing Office; April 2014. Available at: http://www.accessdata. fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=211.80 Accessed September 27, 2014.
- EU
Guidelines to Good Manufacturing Practice, Medicinal Products for Human
and Veterinary Use, Volume 4, Chapter 5: Production, Section 5.20.
Available at: http://ec.europa.eu/health/ files/gmp/chapter5_pc11-2010.pdf. Accessed September 27, 2014.
- USP
<1115> Bioburden Control of Nonsterile Drug Substances and
Products. USP Pharmacopeia Forum, 39, 2013, The United States
Pharmacopeial Convention, Rockville, MD. Available at: http://blog.microbiologynetwork.com/wp-content/uploads/2013/08/394-In-Process-
Revision_-_1115_-BIOBURDEN-CONTROL-OF-NONSTERILE-DRUG-SUBSTANCES-ANDPRODUCTS.
pdf. Accessed September 27, 2014.
- USP <795>
Pharmaceutical Compounding – Nonsterile preparations, Current Version,
The United States Pharmacopeial Convention, Rockville, MD.
- USP
<797> Pharmaceutical Compounding – Sterile preparations, Current
Version, The United States Pharmacopeial Convention, Rockville, MD.
- USP
<1163> Quality Assurance in Pharmaceutical Compounding. Current
Version, The United States Pharmacopeial Convention, Rockville, MD.
- Connors A. Microbial Testing and Monitoring of Compounding Pharmacies. Controlled Environments 2014;17(6):10-11. Available at: http://digital.cemag.us/controlledenvironments/ june_2014#pg10. Accessed September 27, 2014.
- Code
of Federal Regulations, Title 21 Food and Drugs, Parts 600 – 680.
Biologics. Washington, DC: US Government Printing Office; April 2014.
Avaialble at: http://www.accessdata.fda.gov/ scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=600. Accessed September 27, 2014.
- EU
Guidelines to Good Manufacturing Practice, Medicinal Products for Human
and Veterinary Use, Volume 4, Annex 2, Manufacture of Biological active
substances and Medicinal Products for Human Use, 2012. Available at: http://ec.europa.eu/health/files/eudralex/vol-4/vol4- an2__2012-06_en.pdf. Accessed September 27, 2014.
- World
Health Organization (WHO), Environmental Monitoring of Clean Rooms in
Vaccine Manufacturing Facilities, Points to consider for manufacturers
of human vaccines, November 2012. Available at: http://www.who.int/immunization_standards/vaccine_quality/env_ monitoring_cleanrooms_final.pdf. Accessed September 27, 2014.
- PIC/S,
Pharmaceutical Inspection Convention Guide to Good Manufacturing
Practice for Medicinal Products Annex 2, Manufacture of biological
medicinal substances and products for human use, 2014.
- Code of
Federal Regulations, Title 21 Food and Drugs, Part 610.12 Sterility
testing. Washington, DC: US Government Printing Office; April 2012.
Available at: http://www.gpo.gov/fdsys/pkg/CFR- 2012-title21-vol7/pdf/CFR-2012-title21-vol7-sec610-12.pdf. Accessed September 27, 2014.
- Federal
Register, 21 CFR Parts 600, 610, and 800. Amendments to Sterility Test
Requirements for Biological Products, 2012;77(96):26162-26175. Available
at http://www.gpo.gov/fdsys/pkg/FR-2012-05-03/pdf/2012-10649.pdf. Accessed September 27, 2014.
- European
Pharmacopeia, Chapter 7.6, Vaccines for Human Use. Current version,
Quality of Medicines and Healthcare: Strasbourg, France.
- Sutton S. The Environmental Monitoring Program In a GMP Environment. J GXP Compliance 2010:14(3)23-30.
- ISO 14644-1, Cleanrooms and associated controlled environments—Part 1: Classification of air cleanliness 1999. Available at: http://www.iso.org/iso/catalogue_detail. htm?csnumber=25052. Accessed September 27, 2014.
- Lolas
A. The role of microbiology in the design and development of
pharmaceutical manufacturing processes. Pharm. Bioprocess.
2014;2(2):125-128.
- Center for Disease Control. Vaccine
Preventable Deaths and the Global Immunization Vision and Strategy,
2006-2015, MMWR;2006:55(18):511-515
 Zoom In
Zoom In
 Zoom In
Zoom In Zoom In
Zoom In

 Figure 1.
Figure 1.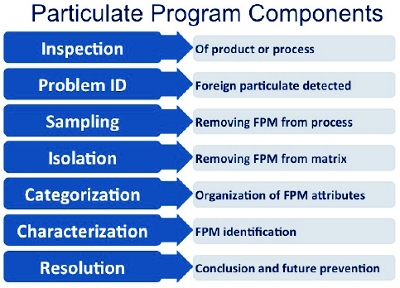 Figure 1. Common Particulate Program Components
Figure 1. Common Particulate Program Components Figure 2. Particles exhibiting similar attributes but of different source materials
Figure 2. Particles exhibiting similar attributes but of different source materials Figure 3. Particulate Characterization Regimen Analytical Techniques
Figure 3. Particulate Characterization Regimen Analytical Techniques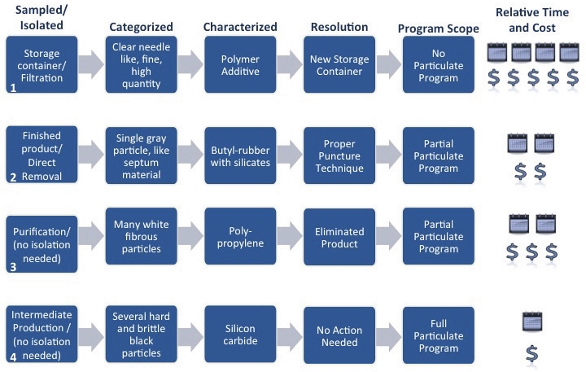 Figure 4. Case Study Summary
Figure 4. Case Study Summary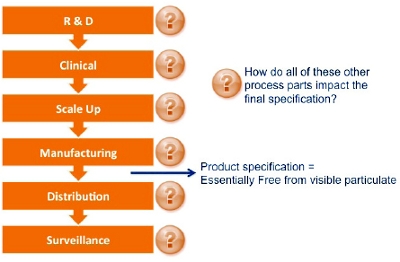 Figure 5. Example Product Lifecycle and End Product Specification
Figure 5. Example Product Lifecycle and End Product Specification
