BY
HERBERT M. SHELTON
Vaccinia
Vaccinia is an acute infectious disease caused by vaccination.
Vaccination is the inoculation of child or adult, well or sick, with septic matter
(pus) derived from suppurating (festering) sores on the abdomen of a previously infected
cow. I think this definition is incomplete in an important respect—I should have said
that it is a criminal operation.
The disease dates from about the year 1774 when an ignorant and
superstitious English farmer, Benjamin Jesty, vaccinated his wife and three children with
matter taken from sores on cows suffering with cow-pox," using a darning needle with
which to make the incisions. Jesty believed a superstition, then prevalent among the
milk-maids, that, one who had had cowpox was immune to small-pox.
Notes of this daring experiment were made by a doctor Nash who died in
1785. At his death these notes passed into the hands of Mr. Thomas Nash who was acquainted
with Edward Jenner, a notorious charlatan, who is credited with having
"discovered" vaccination. In 1789 Jenner inoculated his eighteen month’s
old son with swine-pox matter. He followed this with other inoculations of other children
and the filthy practice of vaccination was definitely launched.
An English writer, Arthur Wollaston Hutton, M. A., says of
Jenner’s framing and qualifications: "But his professional acquirements were but
slender; his medical degree was the outcome of no examination or scientific work,
but merely of a fee of fifteen guineas paid to the University of St. Andrews; while his
other and more important distinction, his Fellowship in the Royal Society, was obtained by
what even Dr. Norman Moore, his latest biographer and apologist, is constrained to admit
was little else than a fraud."
Thus we have a filthy practice, born out of the ignorance and
superstition of the past and fathered by an ignorant imposter and fraud, palmed off on the
world today as a scientific procedure. It is really remarkable, the number of instances in
the history of medicine, of practices and theories now in vogue, that owe their origin to
ancient customs, traditions and superstitions.
It is not known how remote was the belief among the cow hands and dairy
maids of England in the immunizing potency of cow-pox; but it is thought to have come out
of the practice of inoculation which was introduced into England from the East, by Lady
Mary Wortley Montague, wife of the British Ambassador to the Ottoman Court, in 1717. The
practice was abolished by act of Parliament in 1840, due to its evils. In 1754 the Royal
College of Physicians issued the following manifesto, which reads strangely like the
statements made by physicians today about vaccination:
‘The College, having been informed that false reports concerning
the success of inoculation in England have been published in foreign countries, think
proper to declare their sentiments in the following manner, viz.: That the
arguments which at the commencement of this practice were urged against it have been
refuted by experience, that it is now held by the English in greater esteem, and practiced
among them more extensively than ever it was before, and that the college thinks it to be
highly salutary to the human race."
Despite this evident lie by this august body, the practice was not
successful; it was not highly salutary; and experience did not refute the arguments used
against it. It was a very damaging practice which caused an increase in small-pox in
England and was finally abolished by law. Edward Jenner, following Benjamin Jesty, grafted
the old inoculation practice onto the milk-maid’s creed and vaccination (from
vacca—cow) was born.
I mentioned that the inoculation practice was introduced from the east.
The date of the origin of this superstitious practice is hidden in the darkness of
pre-history. Savage and barbaric peoples, in various parts of the world, practiced
inoculation. It is thought to have started in India. where so many of our superstitions
originated, and spread from there to Africa and Europe.
From time immemorial the Negroes and Arabs of Nubia practiced
inoculation against smallpox. The Ashantees and the Moorish and Arab tribes in Northern
Africa practiced arm to arm inoculation from ancient times. Savage tribes of the Upper
Congo practiced it to prevent "syphilis." The Baris of Lado inoculated
themselves over the left breast. The Negroes in Senegal inoculated their children on the
arms. The Moors and Pouls of Senegambia practiced inoculation against pleuro-pneumonia. A
practice of this kind was in vogue in Berne, Switzerland in the 18th century.
The first record of smallpox seems to be in India, where also is the
first record of inoculation, where the practice was in vogue over three thousand years
ago. Dhanwantari, the Vedic father of medicine, and the earliest known Hindu physician,
supposed to have lived 1500 B. C., is said to have been the first to practice inoculation
and it is also stated that the Hindus employed a vaccine. For over a thousand years
inoculation has been practiced in China.
The practice is so mixed up with the religious superstitions of various
peoples that its origin may not be difficult for students of religious history to guess.
In India, in Malaba and in other sections of the world, inoculation was mixed up with the
worship of the smallpox goddess. Inoculation seems to have been nothing more than a
superstitious rite designed to placate and appease the wrath of an irascible deity. People
who imagined all their sufferings were sent upon them because they had offended some of
their gods or goddesses originated the filthy rite to get the goddess into a good humor
again.
According to a Mr. Porter, who was English Ambassador at Constantinople
in 1755 (Gentleman’s Magazine, Oct. 1755): "It is the tradition and
opinion of the country that a certain angel presides over this disease. That it is to
bespeak his favor and evidence their confidence that the Georgians take a small portion of
variolous matter, and, by means of scarification, introduce it between the thumb and fore
finger of a sound person. The operation is supposed to never miss its effect. To secure
beyond all uncertainty, the good will of the angel, they hang up scarlet clothes about the
bed, that being the favorite color of the celestial inhabitant they wish to
propitiate.’
I cannot imagine St. Paul, who refused to eat meat that had been
offered up to idols, baring his arm for pus that is being offered up to the goddess of
smallpox. I cannot imagine Moses, whose Kosher laws, in most of their essential
particulars, are excellent, commanding the Jews to have this trefe stuff inoculated into
their bodies.
Symptoms: Vaccinia begins after inoculation with slight irritation
at the site of vaccination. On the third or fourth day the eruption appears in
the form of a red papule, surrounded by a red areola. On the fifth or sixth day the papule
becomes a vesicle, being filled with a watery substance or a clear substance, with a
distinct central depression (umbilication). By the eighth day the vesicle is perfected and
is then surrounded by a wide reddened zone of inflammatory edema, which is the seat
of intense itching. By the tenth day the contents are purulent (pus) and the
vesicle has become a pustule. The surrounding skin is now much inflamed and painful.
About this time the reddened areola begins to fade and dessication sets in with the
gradual formation of a thick brown crust or scab, which becomes detached and
falls off about the twenty-first to twenty-fifth day, leaving an ugly scar. The
scar is at fist red but gradually becomes paler than the surrounding skin having a punched-out
appearance and is pitted. The evolution of this pathology is accompanied with fever
and constitutional symptoms, malaise, and enlargement of the adjacent
lymph nodes or glands.
Notice the symptoms above described (and this description is gathered
from standard medical works) and you will at once realize that we have been describing an
acute disease—really the acute symptoms of septic infection. Vaccinia will be found
classified in medical books as an acute infectious disease. The infectious matter is pus
taken from pustules on a cow which has previously has previously had pus from the pustules
of a smallpox patient rubbed into incisions in her skin. it is a morbid product, a virus,
and is not and never was "lymph from the calf". Vaccine is pus—it is the
fluid product of suppuration. To vaccinate a person is to produce disease in that person.
It is an effort to prevent disease by producing disease. It does not always "run true
to form." The above description of the disease does not fit all eases.
Complications and Sequelae: Irregular and atypical pocks may
form; several vesicles may coalesce, a general pustular rash, covering the
whole arm or large parts of the body, and called generalized vaccinia, may develop,
about the eighth to tenth day.
Abscess, sloughing. cellulitis, erysipelas, general septic infection,
urticarial eruptions, syphilis, leprosy. tuberculosis, actinomycosis (big jaw), mental
disease, tetanus (lock jaw), paralysis, meningitis, sleeping sickness, etc, may follow. In
rare cases the pock may reappear in the same place after it is apparently healed. In some
instances the abscess that may form refuses to heal. I saw one case of this kind where the
abscess continued to discharge pus after fourteen years. Speaking of generalized vaccinia,
Sir Wm. Osler says: "In children the disease may prove fatal." Osler quotes
Ackland’s arrangements of the dates on which possible eruptions and complications may
be looked for as follows:
"1. During the first three days: Erythema: urticaria; vesicular
and bullous eruptions; invaccinated erysipelas.
"2. After the third day and until the pock reaches maturity:
Urticaria, linchen urticatus; erythema multikormae; accidental erysipelas.
"3. About the end of the first week: Generalized vaccitha;
impetigo; vaccinal ulceration; glandular abscesses; septic infections, gangrene.
"4. After the involution of the pocks: In vaccinated diseases, for
example syphilis."
Under the heading "Transmission of Disease by Vaccination,"
Osler says: "Syphilis has undoubtedly been transmitted by vaccination." Under
the heading, "Influence of Vaccination upon other Diseases," he says: "A
quiescent malady may be lighted into activity by vaccination. This happens with congenital
syphilis, occasionally with tuberculosis . . . At the height of the vaccination
convulsions may occur and be followed by hemiplegia." (Paralysis of one side of the
body.)
Norway suspended vaccination because of hoof and mouth disease being
spread by the practice. In this country our Department of Agriculture traced the epidemics
of foot and mouth disease in 1902, 1908 and 1914 to smallpox vaccine.
It is the medical alibi, when these evils follow vaccination, and they
are far more common than the uninformed may imagine, that they are due to
"carelessness" or to "secondary infections." Dr. Richard C. Cabot
says: "The other thing that bothers people is the fact that vaccination sores get
septic, sometimes when the vaccination is clumsily done, and sometimes when it is
correctly done. We need not necessarily blame the doctor because the patient has a bad
arm. In spite of all precautions, if the patient is in a bad condition, the break in the
skin may become septic."
This is only a half truth. The vaccine sore is septic from the start.
Vaccine is septic matter. Vaccination is deliberate and forcible septic infection. We do
blame the physician, because he introduced the septic matter into the arm.
This picture of vaccination is a black one, but it is by no means the
whole picture. It is almost impossible to exaggerate the evils of this filthy,
superstitious practice and any physician or vaccine propagandist who asserts that
vaccination is harmless is either an ignoramus or a liar. I shall make this quite clear
before I am done with this subject.
"I wish we had known sooner what an awful thing vaccination
is," wrote Mrs A. Kyles, in a letter to the editor of the St. Louis Times, of
Nov 1926, after her boy had died of lockjaw following vaccination. He was vaccinated Oct.
15 and died Nov. 8. 1926; the lockjaw developing about Oct. 31. Thousands of other fond
mothers have cried. "I wish we had known sooner what an awful thing vaccination
is." Why be so willing to believe the sales talks of those who make money out of
vaccines?
On Oct. 7, l926, little Elmer Perry, four-years-old son of Mr.
and Mrs. John Perry. of 35 Schalk St., Newark. N. J. was vaccinated by order of the Health
Authorities. Fifteen days later he become sick, and on Oct. 27 they carried him to the
hospital suffering with lockjaw. A few hours later he died. "They killed my boy, they
killed him," cried the grief stricken father. "They have taken the sunshine from
my life," wailed the frantic mother. This was but one more of thousands of such
tragic scenes. Medical men kill them to save them.
The authorities in this case hastily denied all responsibility for the
boy’s death. They blamed the boy. It is a fair sample of the cowardly manner in which
physicians always disclaim responsibility for their deeds. They are the only class of
criminals of which I know who can escape the penalties for their crimes by placing the
blame on their victims.
On June 20, 1926 little Geraldine Creamer, age 4, 611 John St.,
Peekskill, N. Y.. died of lockjaw, following vaccination during a cooked up smallpox
scare—a case of ivy poisoning having been diagnosed as smallpox. The culprits in this
case explained that the girl, who had been vaccinated on the leg, received the lockjaw
infection from garden soil, while playing in the garden. In a full page article in the New
York Evening Graphic, I challenged them to give me lockjaw by wounding me in a
dozen places and rubbing the soil from the garden in every wound, The Commissioner of
Health made a weak reply in the local paper, but ignored my challenge. He did not want his
alibi exposed by a test,
Lockjaw is a comparatively rare disease except where a vaccination
epidemic rages. In his Principles and Practice of Medicine, Sir Wm. Osler says of
tetanus as a disease transmitted by vaccination: McFarland collected 95 cases practically
all American. Sixty-three occured in 1901, in which R. W. Wilson demonstrated the tetanus
bacillus. Most of these cases occurred about Philadelphia."
The States Public Health Report, March 20, 1925 says that several
fatal cases of tetanus in vaccinated individuals has recently occurred in the United
States" The Report for June 26, 1925, contains accounts, in its first six
pages, of eleven cases of tetanus following vaccination. Boys are more susceptible than
girls to post-vaccinal tetanus.
In a letter dated Aug. 9. 1929, and addressed to Senator Robt. F.
Wagner, Dr. Hugh S. Cummings. Surgeon General of the U.S. Public Health Service, says the
figures which his letter shows are incomplete, for deaths due to post-vaccinal tetanus are
as follows:
1925---29; 1926---15: 1927---17, 1929---1. As most of these deaths
occur after school opens in September, at which time the great orgy of vaccination begins,
the apparent reduction in 1929 is probably very deceptive.
In the early part of 1925, while the whole of the East was in the
throes of a vaccination epidemic, the New York Evening Graphic uncovered at least
two deaths from post-vaccinal tetanus, and many other cases of vaccinal injury in
Baltimore. After they published the accounts of these cases, the hospitals in Baltimore
established a rigid censorship and suppressed the horrid truth about this criminal
practice. But a truce with tetanus; the newspapers carry frequent reports of such deaths
and I can only touch the high spots here. Everyone can know of these cases who cares to
investigate.
Within recent years other troubles have been definitely traced to
vaccination. I have already quoted Dr. Osler’s statement that "at the height of
vaccination convulsions may occur and be followed by hemiplegia." Paralysis is a more
frequent result of vaccination than has heretofore been suspected. Dr. Osler says:
"Cerebro-spinal meningitis has a curious predilection for soldiers." Captain
Sheffield Neave, of England, says, "meningitis is a disease of soldiers and
babies." During World War I there was a great mortality and invalidism among soldiers
due to cerebro-spinal meningitis. Anti— vaccinationists declared it to he due to
vaccination. This brought vigorous protests and loud denunciations from the devotees of
pus and the smallpox goddess.
In the "Lancet" the leading British medical Journal, of
September 4. 1926, is set forth accounts of seven cases of encephalo-myelitis
(inflammation of the brain and spinal cord and their membranes), following vaccination in
two London hospitals within recent years. Prof.. H. M. Turnbull and Prof. Jas. McIntosh
who painfully and carefully investigated these cases stated in the British Journal of
Experimental Pathology, from which the "Lancet" quotes, that: "There
can be no doubt that vaccination was a definite causal factor."
The Lancet declares that the account in the Journal:
"includes summaries of clinical histories and necropsies and descriptions of the
pathological changes, gross and minute in the central nervous system as well as in the
vaccinated areas, regional lymphatic glands, and other tissues. Beautifully clear drawings
illustrate the histological lesions found in the spinal cord at lower levels. The evidence
of aetiology (the science of cause) derived from clinical and histological manifestations
is shown to be strong, and is confirmed by the results of biological experiments
(experiments on animals made independently by Dr. Paul Fildes and Prof. McIntosh).
Inoculation of material from the brain and spinal cord of three of these cases showed the
presence of vaccinia virus, no other virus being obtained."
The suspicions of these physicians were first aroused in 1912 when a
post-mortem on a recently vaccinated boy of 15 years revealed encephalo-myelitis. In
December, 1922, a 9 year old girl came to necropsy with a diagnosis of tubercular
meningitis. However the microscope revealed no lesions except recent vaccination scabs,
glandular inflammation, in the region of vaccination and slight changes in the central
nervous system. Brain and cord presented the same peculiar changes as those found in the
boy ten years previously.
"Other cases," says the Lancet, "were now quickly
recognized one in a man of 21, and ‘the rest in girls of 7, 12, 15, and 22
respectively. All these patients except one girl died in the course of an acute attack of
encephalo-myelitis complicated by broncho pneumonia."
As an example of how these seven cases proceeded the case of the woman
22 years of age will suffice. She was vaccinated while an infant and again on November 29,
1922. Seven days thereafter she developed a severe headache and other symptoms. On the
10th and 12th days she was drowsy and had high fever, On the 13th day she became
semi-comatose and on the 14th day she died.
The Lancet for October 9, 1926, states that in Holland, during
the period from January 1, 1924, to July 1, 1925: "35 cases, of which 15 were fatal,
occurred of Encephalitis following vaccination after an interval of 10 to 30 days,"
had elapsed.
he Lancet further declares in the article previously quoted
from: "Investigation of the possible path of infection gave negative
results—Close examination of the vaccinal areas and regional glands yielded but
little information, since the histological changes appeared to be essentially similar to
those in a control case, a recently vaccinated boy killed in an accident."
This means that the ordinary and regular course of mischief pursued by
vaccination may easily result in the production of these diseases. The Lancet further
says: "Though the path of infection cannot he traced, the authors would appear to
have ample justification for concluding, in view of the close resemblance between the
clinical histories, the uniformity of the pathological findings, and the absence of
similar cases independent of vaccination that vaccination was a definite causal factor and
no chance coincidence." (Italics mine).
In the year 1927 when Mr. Marky and Senator Love debated on
vaccination, we exhibited on the platform, a little girl whose body was frightfully
twisted, greatly emaciated and paralyzed as a result of vaccination. With the smooth
sagacity of the suave politician and with resort to the ancient medical subterfuges of
"secondary infection" and "intercurrent malady," Dr. Love attempted to
make the audience believe the child’s troubles were due to something other than
vaccination. But an "intercurrent affection" is mere bunk. It never existed
outside the medical mind. The Lancet had formerly held to the same theory with
regard to such cases as cited above. Referring in its issue of August 1, 1925, to the
numerous cases on the continent, it declares: "Experiment and pathological research
have shown that this form of the disease is not due to the virus of Jenners
vaccine"…."There was latent infection" and "vaccination merely
hatched it out."
"Latent infection" is another subterfuge that has long
served the blundering medical profession when tuberculosis, syphilis and leprosy follow
vaccination. But the end of this subterfuge is drawing near. The Lancet has unsaid
what it declared in the quotation above. It declares: "Similar cases independent of
vaccination were not observed at the same time nor any other time. The authors give cogent
reasons against the assumption that the post-vaccinal cases described by them and by
workers abroad are merely examples of poliomyelitis, (inflammation of the gray matter of
the spinal cord) or encephalitis lethargic a (sleeping sickness), in which vaccination was
an immaterial accident." It declares that encephalo-myelitis following vaccination
always exhibits more extensive lesions than those of sleeping sickness and that
"histologically, the inflammation in ordinary cases of poliomyelitis (infantile
paralysis) differs conspicuously from that following vaccination." In 1923, 1924 and
1925 great efforts were made in England to have everybody vaccinated. Thousands of
vaccinations were performed. There occurred a great increase in the cases of
Encephalitis-Lethargica. In 1924, there were 6,296 cases of this and similar affections
reported in England and Wales, with a population of 38,746,000; or 162 cases per million
of population. In Liverpool, with a population of 836,000 there were reported 257 such
cases; or 306 cases per million of population. Liverpool was fifty per cent better
vaccinated than the average of England and Wales, and had almost 100% more Encephalitis. I
presume this was due to an "intercurrent affection," or a "latent
infection," or to a "secondary infection."
The New York State Journal of Medicine, May 15, 1926,
carried two articles from foreign Journals discussing similar cases on the European
continent. In one of these Carl Leiner, (Vienna) is said to have discussed encaphilitis
and meningitis developing in nine to fifteen days after vaccination. He admits that in a
generalized infection, like generalized vaccinia, there may be intracranial complications.
The article also states that Dr. Lucksch saw three cases and knew of four more and of the
seven children, five died. In two autopsies, which he obtained, he was able to show beyond
doubt that "death had been due to encephalitis." Bastianse, of the Hague,
collected notes of 34 similar cases which occurred in Holland during 18 months of 1924-25,
with a mortality of forty per cent—"deadlier if anything than ordinary epidemic
encephalitis." "In addition several cases of serious meningitis have been
reported."
Three cases reported,, by the author of the article, in Austria, showed
that "not only the encephalon but the cord and peripheral nerves may be involved, s
that the affection may be spoken of broadly as a meningoencephalitis polyneuntis."
The other article is a brief of an article by Dr. W. F. Winkler, chief
of the University Clime of Rostock. It says: "Quite recently isolated cases of
cerebral symptoms, suggesting encephalitis, following vaccination have been reported from
Holland, Czechoslovakia, and Germany and from Switzerland there have been reported two
cases of serious meningitis."
The Netherlands, and other countries, for instance, France, have also
reported cases of this kind. In the Journal of the American Medical Association, July
3, 1926, p. 45, is an article by its Berlin correspondent discussing "Nervous
disturbances and Smallpox Vaccination." In it are these words: "In regions in
which there is no organized vaccination of the population, general paralysis is rare. In
patients with general paralysis he (Dr. Daraskwiewicz), has never seen smallpox scars, but
vaccination scars were always present." Physicians of Holland declared: "It is
impossible to deny a connection between vaccination and the encephalitis which follows
it." It is noted that, whereas, boys are most susceptible to post-vaccinal tetanus,
girls are most suceptible to post-vaccinal encephalitis.
It would be idle to assert that all cases of local or general paralysis
are due to vaccination. There are cases due to other causes also. But these other cases
must not be made a basis for denying the evil influence of vaccination, as some vaccine
apologists attempt.
How new is the phenomenon? Who knows? Dr. Pierre Baron, Ancien Intern
of the Hospitaux of Paris, prefaces his work on post-vaccinal encephalitis (1929), in
which his conclusions are based on his own observations, with a case he found after
searching through medical annals and unearthed a report of a case in the "Archives
de Medicine des Enfants," in 1907. Dr. Combay of the Medical Society of
the Hospitals of Paris, reported a case which had occurred in his practice in 1905. Dr.
Comby tells of a baby girl, in excellent health when vaccinated at four months of age, who
developed convulsions on the eighth day, followed by strabismus and other troubles. She
did not die but was left with an "important sequel." She no longer recognized
her surroundings; almost forgot how to nurse; had a vague look; "veritable
intellectual obnubilation," developed idiocy with progressive cerebral sclerosis
(hardening of the brain), and nearing her eighteenth month died. Her death went into
medical "statistics" as due to pneumonia—an old trick in hiding their
crimes.
Dr. Baron’s book discusses 255 cases of post-vaccinal encephalitis, avowedly
discussed as such in medical works. His list is far from complete, for he credits the
United States with only four cases, all of these before 1927.
Great Britain appointed two committees to investigate this
matter—the Andrews Committee, appointed Nov. 1923, which made its report May 1925;
and the Rolleston Committee appointed Feb. 1926, which made its report Feb. 1928. These
two committees were composed of eminent medical men all of whom supported vaccination.
The Andrews Committee reported 62 cases of post-vaccinal encephalitis
with 36 deaths—40 females and 22 males; average age 10 1/2 years. Four cases were
under one year, one case fifty years, and forty-eight cases were from six to sixteen
years. Government vaccine had been used in 53 of these cases, of which 30 were fatal. The
Rolleston Committee reported 30 cases with 16 fatalities. Government vaccine was used in
18 of these with 8 deaths. This committee also reported the subsequent history of 10
non-fatal cases under 15 years, showing that 4 were permanently injured in some
way—in mind, memory, temper, vigor, relapse.
Since vaccination was made compulsory in England and Wales one million
infants have died (to 1930) of convulsions, tetanus, encephalitis, meningitis, and other
nervous ailments. How many of these were due to vaccination there is now no means of
knowing, but in the light of present facts, we are safe in assuming that a large
proportion of them died from this cause.
In 1924 there were recorded in England and Wales 5,039 cases of
encephalitis lethargica, 397 of cerebro-spinal fever, 777 acute poliomyelitis, 83 of
polio-encephalitis—a total of 6,296 cases, with 2,200 deaths, 2,520 permanently
injured brains (insane), and 1,575 complete recoveries. The cases in 1924 were three times
as great as the yearly average for the nine preceding years. In 1922-23-24 the physicians
of England and Wales cooked up a number of smallpox scares causing 288,000 revaccinations.
"Extra vaccination was followed by this extra crop of sleepy sickness."
A case of post-vaccinal encephalitis was reported in Ireland in 1930 in
a baby boy of 10 pounds. He was vaccinated on May 3 and became ill on May 10, "being
cross and very restless with vomiting. Next day he was quiet and apathetic and on
admission to the hospital his condition resembled tetanus."
The League of Nations in its Report of Aug. 27, 1928 mentions 139 cases
and 41 deaths in Holland. This resulted in Holland stopping compulsory vaccination during
1928-29. The total number of vaccinations in Holland in the first half of 1928 was less
than one-third of those for the first half of 1927 and the deaths from encephalitis were
reduced to less than one-third.
Germany modified her compulsory vaccination law. She adopted an
optional clause, such as the one England had. The International News Service, Feb.
27, 1930 informs us: "The change of attitude of some medical experts towards
vaccination in favor of a less rigid enforcement of the law has been brought about mainly
through a considerable number of post-vaccinal diseases observed in Holland and England
and in sporadic cases in Germany.
"Vaccinated people developed a sort of cerebral inflammation
(encephalitis post-vaccinalis) which resulted in a number of deaths and in several cases
of a mild form of mental derangement."
Here is part of an item which appeared in the Journal of the
American Medical Association for April 5, 1930: "Reisch reports that following
the vaccination of 233 children aged between 5 and 10 years, several cases with
encephalitic symptoms were observed. Two were especially severe and ended fatally. The
necropsy revealed the changes characteristic of encephalomyelitis. Six other children also
developed encephalitic symptoms from six to twelve days after the vaccination."
The Report of the Commission of Smallpox and Vaccination of the Health
Organization of the League of Nations, Geneva, Aug, 27, 1928, says: "The
post-vaccinal encephalitis with which we are dealing has become a problem in itself mainly
in consequence of the events of the last few years in the Netherlands and England and
Wales. In each of these countries the cases which have occurred have been sufficiently
numerous and similar to require them to be considered collectively. Their occurrence has
led to the realization that a new, or at least a previously unsuspected or unrecognized,
risk attaches to the practice of vaccination."
Now what of America? Do such cases ever occur here? They do. But they
are seldom reported and, it seems, are never investigated. In 1930 Julia Motley, age 12,
of Irisburg, Va., died of acute infantile paralysis which "seized" her 3 weeks
after she had been vaccinatëd. Her parents attributed her death to vaccination, whereupon
the State Health authorities came to the rescue of vaccination. The News Leader, Richmond,
March 28, 1930 says: "While the parents gave vaccination as the cause of death, Dr.
J. V. Shackleford, the physician, states that the death certificate (made out by him, of
course), shows that the little girl died of acute infantile paralysis, with which she was
seized three weeks after she had been vaccinated."
And that’s that! The physician who vaccinated the girl makes out
the death certificate to shield himself and the vaccine and the matter is settled. The
girl is now immune to smallpox and the smallpox goddess has been appeased. This reminds me
very much of a statement contained in the memorandum, of Professor Jorge, to the Committee
of the International Office of Public Hygiene (published in the monthly bulletin of that
organization, for Jan. 1927) where he refers to "the motives which weighed with us
not to noise abroad in the great press the news of this complication of a prophylactic
operation hitherto looked upon (sic) as innocuous . . ." (Italics mine.)
The press probably would not have published the news had they given it
out, for, it always protects the medical profession. The press is as good about
suppressing the truth as Professor Jorge and his coworkers. The mediums of intelligence
(?), our newspapers, magazines, movies, churches and schools, play a vast part in the
continual bunking of our more or less brainy public, while every subsidized press or
scientist. professor or preacher, is entirely a political organ, at the heck and call of
the exploiters. Of course, when it is all said and done, the class of nincom-poops who
take any stock in the stuff dished out, do not really count. They are like the defenders
of any kind of "it-works-one-day-a-week" philosophy: in that when the tide
rises, they will be found to be without a bathing suit.
Surgeon Chas. Armstrong, in Public Health Reports, Aug. 23,
1929, says in an article on post-vaccinal encephalitis: "In so far as the age factor
is concerned, the custom in this country of performing primary vaccinations at the sixth
or seventh year would seem to predispose our population to the complication. Cases have,
moreover, occurred. Wilson and Ford, and Fulgham and Beykirk have reported 3 cases in this
country which were confirmed by pathological studies. Other possible cases based on
clinical and epidemiological grounds have been reported from Connecticut, Rhode Island,
New York, Maryland, Illinois, California. Washington. and the District of Columbia."
The Weekly Bulletin of the Dept. of Health, of New York City,
Sept. 7. 1929 devotes several pages to a discussion of post-vaccinal encephalitis
and says: "Although only a few cases have been reported in the United State, it seems
advisable to call physicians’ attention to this complication so that any cases in
which persons recently vaccinated show symptoms pointing to the central nervous system can
he carefully investigated."
It may be interesting enough for physicians to study symptoms pointing
to the central nervous system but it will not be interesting to you or your afflicted
child. Since the medical profession is determined not to abandon this filthy and deadly
practice, no matter how many children are sickened, maimed and killed, it is up to you to
prevent post-vaccmal encephalitis, and all the other troubles discussed in this chapter,
by not permitting sour child to be infected with this dirty cow pus.
It is your child. It does not belong to the state. It was not born into
this world to furnish money to the medical profession. You are responsible for its care
and training. If you betray your child by giving it over to this modern moloch, you
deserve a worse fate than any Dante ever pictured. Parents owe certain responsibilities to
their children. One of these is certainly to guard these children against attack from all
foes, including the foes of their health. It is the duty of every parent to "refuse
and resist" vaccination for his or her children, wherever such a parent may live and
whatever the circumstances under which the vaccination is demanded. Fight, go to jail,
resist in every possible manner the cow-pox bullies and their putrescent points. In Italy
some years ago, when a group of physicians invaded the homes of Italian mountaineers to
forcibly vaccinate the children, the mountaineers simply stripped the pus-punchers of
their clothes, gave them a liberal dose of their own "medicine," and sent them
scurrying home. I recommend this measure for immediate adoption in this country. Let the
rascals suffer as they make others suffer. It will teach them a much needed lesson.
In reply to an inquiry, addressed to the United States Public Health
Service, by Senator Robt. F. Wagner, New York, Surgeon Ceneral Hugh S. Cumming says:
"One case (of encephalitis followirig vaccination) in the United States was published
in 1929 and two in 1927. These three cases seem to be definitely established as sequebe of
vaccination. Several other cases less well established have come to our attention but need
not be considered here."
That these and all figures given in this reply are not complete is
evident from the closing paragraph of his letter. He says: "Although a search has
been made of the literature since 1925, we cannot be sure that this is a complete list.
While the Public Health Service endeavors to learn of and in many instances to investigate
untoward cases suspected of being caused by biologic products, there is no legal mechanism
requiring the reporting of such cases to the Public Health Service."
The Report of the Surgeon General of the U. S. Army, 1918, shows that
during 1917 there were admitted to the army hospitals 19,608 men suffering from
anti-typhoid inoculation and vaccinia. The Report for 1919 covering the year 1918 shows
the total admissions suffering from typhoid vaccination to be 23,191, and 10,830 suffering
from vaccinia. Assuming that the proportions of those suffering from these two
inoculations were about the same for the two years it means that approximately 20,000 were
in the army hospitals admittedly suffering from smallpox vaccination. This takes no
account of those whose sufferings were attributed to something else, nor of those whose
sufferings; though great, were not great enough to cause them to be sent to the hospitals.
The Chicago Tribune, June 6, 1926, carried the account of the
death of Kasmir Jeskey, 10-year-old son of Mrs. Anna Jesky, 1523 17th Ave., Meirose Park.
The Tribune stated: "Blood poisoning believed to have resulted from
vaccination yesterday claimed the life of Kasmir Jesky."
The Report of The Register General, England, from 1875 to 1923
recorded 1,464 deaths officially admitted to have been caused by vaccination. These
figures give but a small part of the picture for most such deaths are covered up. For
instance, in one series of deaths caused by vaccination, Public Enquiry revealed that
vaccination had been mentioned as a cause in only one case. In another series of seventeen
deaths following vaccination, investigated by a medical man, who published the details,
only one death had been attributed to vaccination. One British physician said: "In
certificates given by us voluntarily and to which the public have access, it is scarcely
to be expected that a medical man will give opinions which may tell against or reflect
upon himself in any way, or which are likely to cause annnoyance or injury to the
survivors. In such cases he will most likely tell the truth, but not the whole truth, and
assign some prominent symptom as the cause of death. As instances of cases which may tell
against the medical man himself, I will mention erysipelas after vaccination and pueperal
fever. A death from the first cause occurred not long ago in my practice, and although I
had not vaccinated the child, yet in my desire to preserve vaccination from reproach, I
omitted all mention of it from my certificate of death."
Vaccination must be saved from reproach at all costs. Who cares how
many children are killed if only vaccination may be saved from dishonor. It is up to
parents to put an end to this crippling and maiming of children. It is the sacred duty of
all parents to protect their children from all harm. If the medical profession is not
honorable enough to abandon this highly remunerative, though evil and deadly practice, it
behooves parents to cut their professional throats.
Will it be urged that while vaccination is often productive of harm and
death, it produces less of these than it prevents? If so, I shall show that this is not
true. But, grant for a moment the truth of the assertion, it is still true that to force
such a dangerous process upon one is unjustifiable. It is a danger and we each have a
right to choose between two dangers. Compulsory vaccination is a crime.
The Christian Herald, England, July 7, 1927, carries an account of
a smallpox epidemic, of a very serious type, in 15 departments (counties) in France, with
a death rate of nearly 50 per cent in women and about 33 per cent in men. All of these
cases were vaccinated people—many of the victims having been vaccinated as many as
three times. If vaccination protects, why did it fail in these cases?
In our army during the Spanish American War and in the Philippines the
soldiers had been vaccinated, not only annually, but every six weeks. Chief Surgeon
Lippincott said: "Vaccination is carried on as regularly as post drill." Yet the
official report shows 276 cases of smallpox in 1899 with 78 deaths; 246 cases in 1900 with
113 deaths; and 125 cases with 37 deaths in 1901; the case fatality of nearly fifty per
cent, in 1900 being the highest ever recorded for this disease in the army a well
vaccinated army, if ever there was one.
In 1872 Japan passed a compulsory vaccination law which was rigidly
enforced. But smallpox continued to "ravage" that country. In 1885 another law
was passed requiring revaccination every seven years. From 1886 to 1892 there were
25,474,370 vaccinations, revaccinations and re-re-vaccinations recorded in Japan. During
these same seven years, 1886 to 1892, Japan had 156,175 cases of smallpox, with 38,979
deaths, or a case fatality of nearly twenty-five per cent which exceeds the smallpox
death-rate of the pre-vaccination period when nobody was vaccinated. In a single year
(1893) Japan had 41,898 cases of smallpox with 11,852 deaths.
In 1896 the Japanese Parliment passed an act, which was immediately
signed by the Mikado, requiring every resident of Japan, whatever his or her station in
life, to be vaccinated and revaccinated every five years. The act was rigidly enforced
under severe penalties. Baron Takalira boasted in London in 1906, at the Jubilee Dinner of
the Society of Medical Officers of Health of England that:
"There are no anti-vaccinationists in Japan. Every child is
vaccinated before it is six months old, revaccinated when it enters school at six years
and again re-vaccinated at fourteen years of age when going to the middle school, and the
men are re-vaccinated before entering the army, while a further re-vaccination is enforced
whenever an outbreak of smallpox occurs."
Notice the last part of this statement. If vaccination prevents
smallpox, how do "outbreaks of smallpox" occur in such a thoroughly vaccinated
country? There can be but one answer; namely, Vaccination does not protect.
This compulsory vaccination law became effective in Japan in 1896. In
1897 there were 49,946 cases of smallpox in Japan, with 2,276 deaths from this cause. In
1908 there were 10,067 cases with 5,837 deaths officially recorded. From 1889 to 1908
Japan had 171,611 cases of smallpox with 47,919 deaths. If anybody thinks that
vaccination, re-vaccination, and re-re-vaccination prevents or mitigates smallpox, let him
look at these figures. Here is a case fatality of nearly 30 per cent. It would be
interesting to know to what extent the disease was mitigated by vaccination in
those 47,919 fatal cases of post-vaccinal smallpox.
The New York Medical Journal, July 22, 1899, contains an article
on "Vaccination in Italy," by Chas. Rauta, M.D., Prof. of Hygiene and Materia
Medica in the University of Perguia, Italy. In this he points out that "Italy is one
of the best vaccinated countries in the world, if not the best of all, and we can prove
that mathematically." He says further: "For twenty years before 1885, our Nation
was vaccinated in the proportion of 98.5 per cent. Notwithstanding, the epidemics of
smallpox that we have had have been something so frightful that nothing before the
invention of vaccination could equal them." "During 1887, we had 16,249 deaths
from smallpox; in 1888, 18,110; and in 1889, 13,413."
Referring to the Italian army, in which "vaccination had been
performed twice a year in the most satisfactory manner for many years past" he says
that, "now we see that soldiers not protected because vaccination did not
‘take’ were less attacked by smallpox than those ‘duly protected,’ by
the good results of their re-vaccination; and that the death-rate in those vaccinated with
good results was greater than among those in whom the vaccination did not take."
We have forced vaccination on the Philippines since we took over the
Islands. Spain had done the same thing previously. In 1905-06; 1907-08 and in 1918-19
these Islands experienced severe smallpox epidemics, the 1918-19 one being the worst of
all. There were 47,887 cases of smallpox with 16,578 deaths officially reported in 1918.
In Manila alone, the best vaccinated part of the Islands, there were 1,326 cases and 869
deaths, or a case mortality of 65.3 per cent. The lowest mortality, 11.4 per cent was in
Mindanao, the least vaccinated portion of the islands.
The Health Service got busy and vaccinated thousands and thousands,
performing about four vaccinations for each inhabitant in Manila. This was followed in
1919 with 99,300 cases of smallpox, with 47,395 deaths. In two years time in a population
of less than 11,000,000 there were 147,187 cases of smallpox and 63,973 deaths.
The 1920 Report of the Philippine Health Service, (see pages 141 and
142), makes the following very brief comment: "From the time in which smallpox was
practically eradicated in the city of Manila to the year 1918 (about 9 years)
in which the epidemic appeared certainly in one of its severest forms, hundreds after
hundreds of thousands of people were yearly vaccinated with the most unfortunate result
that the 1918 epidemic looks prima facie as a flagrant failure of the classic
immunization towards future epidemics." (Italics mine.)
Alibies were offered for the failure, however, and the dirty work
continues. No matter how great the evil, those who profit from it will not correct
it—not so long as profits are still to be made therefrom.
There is an unvaccinated country in this world without smallpox.
Australia is the great unvaccinated country and despite dire predictions of disaster from
vaccine advocates, Australia remains free from smallpox. Three-fourths of her population
have always been in the never-vaccinated class. Under the modern theory that vaccinal
immunity lasts only five years (Italy vaccinated twice a year and failed) 21/2% of her
population are "protected."
In the whole of Australian history less than one person a year has died
of smallpox. Many of these were from the outside and were simply quarantined there. In
Queensland where the official figures show 1 vaccination for every 1,500 births the state
has had but one "outbreak." In 1892 a well-vaccinated quarantine official
"contracted" the disease on ship. There were no other cases. The
"epidemic" had no show among an unvaccinated people. In Victoria in 21 years
there were 5 deaths from smallpox and 14 deaths from vaccination—these are only those
deaths that are honestly attributed to this cause. This coincides with the reports of the
Register General of England covering a period of years in which there were, in England, 42
deaths from smallpox under five years of age and 157 deaths officially admitted to have
been due to vaccination. There are also the official figures which show that "only
109 children (under five) in England and Wales died of smallpox in the twenty-nine years
ending December, 1933, but 270 died of vaccination" in the same period in these two
countries.
In England and Scotland the decline of vaccination has been accompanied
by the practical disappearance of smallpox. Here are the figures, briefly England, 1871-75
percentage of vaccination 97.6%; smallpox deaths per million people, 228; 1910-20
percentage of vaccination 43.9; smallpox deaths per million people 0.4.
Scotland, 1855-1874 one of the best vaccinated countries of the world,
"not an unvaccinated child in Scotland;" 9,087 children under five years old
died of smallpox; 1907-1919 with about one-third of the children vaccinated only 7 deaths
under five years from smallpox.
I would not go so far as to say that vaccination has never saved a
single person from smallpox. It is a matter of record that thousands of the victims of
this superstitious rite have been saved by the immunizing potency of death. But it
is a fact that the official statistics of England and Wales show unmistakably that, while
vaccination has killed ten times more people than smallpox, there has been a decrease in
smallpox concomitant with the decrease in vaccination. The following table of official
statistics from England and Wales giving the average annual percentage of births
vaccinated and the number of smallpox deaths registered will prove instructive to all
intelligent readers:.
Period
Percentage of births
Vaccinated Smallpox deaths
1872-1881
85.5
3,708.2
1882-1891 .
82.1
923.0
1892-1901
67.9
436.5
1892-1911..
67.6
395.3
1912-1921
43.3
12.2
1922-1931
43.1
25.0
1932-1941
34.9
1.4
During the period when 85.5 percent of all babies born were vaccinated
and another ten per cent of them died before they were old enough for vaccination, these
two countries had an average annual number of 3,708 deaths from smallpox. When vaccination
had declined until only about one-third of the infants born were vaccinated the average
annual death rate in smallpox had dropped to less than two a year. It may be appropriately
asked, in the words of the Vaccination Inquirer (London), Feb. 1947: "How
could an operation that was declining be responsible for the extermination of
smallpox?"
In 1942 a case of smallpox at Swindon (Britain) resulted in the
vaccination of large numbers of people. Only three cases of smallpox occurred and these
all recovered, but twelve vaccinated individuals died from inflammation of the brain. In
the same year near Edinburg, Scotland eight people died of smallpox (six of these had been
vaccinated) while ten died from the effects of vaccination.
In Britain during the years 1939 to 1944 there were 60 cases of
post-vaccinal encephalitis, 31 of whom died. This is a fatality rate of slightly more than
fifty percent. During this same period, there were but 21 cases of confirmed smallpox with
but three deaths. There were, in other words, in Britain during this period, three times
as many cases of post-vaccinal encephalitis as of smallpox and ten times as many deaths
from post-vaccinal encephalitis as from smallpox. As these figures are official and are
supplied by the British medical profession itself, as they make the diagnoses and report
the cases and deaths, they constitute damaging admissions by the profession that, while
vaccination does not prevent smallpox, vaccinia is a much more dangerous disease than
smallpox.
Since the first edition of this book was published, England, the first
nation in the world to pass a compulsory vaccination law, has repealed her law and no one
in the British Empire, not even in her armed forces, is forced to submit to vaccination.
Since 1907 nobody among her citizens or in her armed forces was compelled to be vaccinated
if he conscientiously objected to it. On the same ground parents could avoid vaccination
for their children. In this land of the craven and home of the slave, a land that proudly
boasts that it gained its freedom in 1776 from this same British Empire, vaccination is
still compulsory in several of our states and in many cities outside these states, as well
as in the armed forces of the country. Civil service employees are also compelled to
submit or lose their jobs.
Smallpox is always worse where vaccination abounds. The scratch of
vaccination is the "scratch of death." Yet our medically controlled Health
Boards cook up fake epidemics, create panics for profit, such as the ones in Kansas City
in 1921, Pittsburgh in 1924, Philadelphia, Baltimore, Washington in 1925. An effort was
also made to create a panic in New York in 1925, but due to the open fight against it by
the New York Evening Graphic, the Commissioner of Health called it off.
Surgeon J. P. Leake, says in Public Health Reports, Jan. 28,
1927, the weekly bulletin of the U. S. Public Health Service: "Will a nonimmunized
person contract smallpox if exposed to the disease? By no means uniformly. Exposure to
smallpox, especially to the milder forms, without contracting the disease frequently
occurs and is no definite evidence of immunity. The number of cases of smallpox among the
unprotected persons in contact with patients suffering from the disease is very much less
than 100 per cent
"Though smallpox is unquestionably many times more frequent in the
unvaccinated than in those who have had even a single vaccination, it is believed that
neither the vaccination history nor the presence of scars should be given diagnostic
weight. The unreliability of such a criterion is especially evident in virulent outbreaks
of the disease
"The purpuric, uniformly fatal form of smallpox , is the most
difficult to prevent by vaccination, and cases of this form, without a true smallpox
eruption, may occur in persons with a fairly good vaccination history . .
"The mildness of the form of smallpox commonest at present is one
reason for endeavoring to make preventive vaccination as harmless and as mild as possible
"Cases and even fatalities, occur in every severe epidemic among
persons who were vaccinated in good time but with vaccine found, too late, to be of
insufficient potency; such cases and fatalities also occur among persons thought to be
protected by successful vaccination performed years previously."
You are vaccinated and have smallpox. The vaccine was of
"insufficient potency," although this was discovered too late—that is,
after you have the smallpox. You are vaccinated and do not develop smallpox—it is
assumed that the vaccine was potent. It is like the old test for mushrooms—eat them
and live they are mushrooms; eat them and die, they are toad stools.
In 1926, 130 members of the Dallas (Tex.) Chamber of Commerce cancelled
their trip to Mexico because vaccination was required as a precedent to entrance. Nearly a
100 medical men, at a conference in Dallas, went to Mexico, after they obtained permission
to enter without being vaccinated. Think this over before you submit your child to this
evil and superstitious rite.
In this country the risks from vaccination, according to official
figures, which are slanted in favor of vaccination, is ten times greater than the risk of
smallpox. According to the figures of the United States Public Health Service there were
officially reported an average of sixteen deaths per million vaccinations in this country
in the years 1925 to 1928 inclusive. These deaths cover only those officially admitted to
have been due to vaccination and do not include the deaths from encephalitis, meningitis,
etc., which resulted from vaccination. Up to that time twenty cases of encephalitis had
been officially reported in the United States as resulting from vaccination. The Public
Health service instituted an investigation to determine the extent of such cases in this
country, but I have seen no report of their findings.
During the years 1927 to 1929 inclusive there were officially reported
an average of 1.18 deaths per million population from smallpox in the United States. In
many of these deaths, smallpox is not given as the primary cause of death. Deaths from
smallpox have almost reached the vanishing point in this country, only thirteen states of
which require vaccination precedent to school attendance and none of which require
vaccination of infants and adults outside the armed forces.
Due to the fact that vaccination is more dangerous than smallpox, many
leading medical men refrain from vaccinating their own children. An editorial in American
Medicine, March, 1914, says:
"The growing opposition to vaccination is a matter of grave
concern. This new movement . . . is not the illogical and absurd anti-vaccinationist
crusade, but is the conviction on the part of very intelligent men, that it is useless to
protect against an infection which they may never encounter . . . This attitude is not
confined to laymen, but is taken by those leading men in the medical profession who
postpone vaccination of their own kith and kin until the last moment. Two world renowned
men have confessed to us that they have had their children vaccinated only in obedience to
public opinion in and out of the profession . . . So we hear men saying that there is not
one chance in a million of their children being infected with smallpox, but that there is
far more chance of pus infection or tetanus from the vaccine .
This criminal practice will end as soon as parents develop sufficient
interest in the welfare of their children. At present parents offer up their children on
the altars of the smallpox goddess, because commercial ghouls demand it, and hope that the
children will not be greatly injured. If a child is invalided for life or is killed, the
parents meekly accept the lying excuses of the scoundrels who maim and murder children for
money, cry a little, and return to their movies and joy rides. Reader, do you know how
Judas felt after he had sold his master for a few pieces of silver? If you have
surrendered your child to be vaccinated and inoculated, after you learned the truth, you
know how he felt. There is one great difference between you and him—Judas had decency
enough to go out and hang himself.



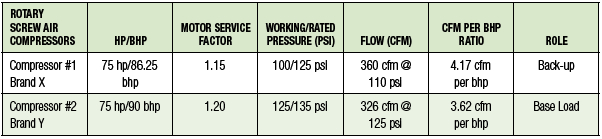

 igation
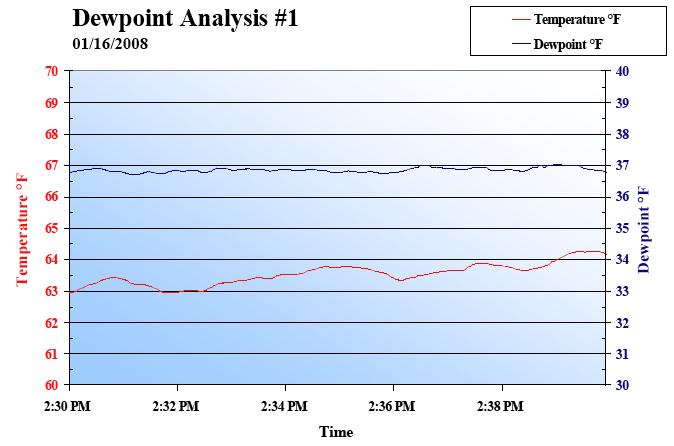
we found the root of the problem. When air is discharged from the
piping between the cylinder and the valve, the temperature of the air
drops due to adiabatic expansion. . If the atmospheric dewpoint of
the supply air is T1, and the temperature of the air T2 after adiabatic
expansion falls below this value (T1
igation
we found the root of the problem. When air is discharged from the
piping between the cylinder and the valve, the temperature of the air
drops due to adiabatic expansion. . If the atmospheric dewpoint of
the supply air is T1, and the temperature of the air T2 after adiabatic
expansion falls below this value (T1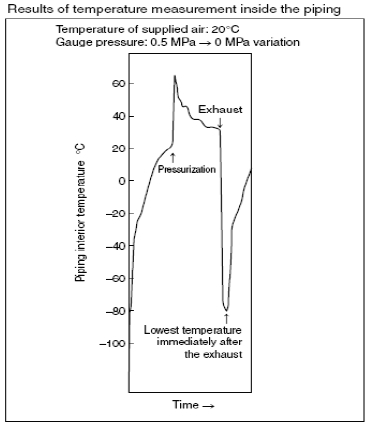





 The
FDA has taken notice, of course, and the quality of the air being used
is a concern; and, rightly so, no standard has been issued for the use
of compressed air in production.
The
FDA has taken notice, of course, and the quality of the air being used
is a concern; and, rightly so, no standard has been issued for the use
of compressed air in production.
 The
manufacture of pharmaceutical, bio-pharmaceutical and medicinal
products is unique from many other manufacturing processes for a number
of reasons. The most important is that these products are utilized for
the health and well being of living organisms, including humans.
Because of this, the design of the manufacturing area of these
facilities is required to follow Good Manufacturing Practice (GMP).
The
manufacture of pharmaceutical, bio-pharmaceutical and medicinal
products is unique from many other manufacturing processes for a number
of reasons. The most important is that these products are utilized for
the health and well being of living organisms, including humans.
Because of this, the design of the manufacturing area of these
facilities is required to follow Good Manufacturing Practice (GMP).