Biotherapeutic medicines generated by living cells or organisms are larger and more complex than chemically synthesized, small molecule medicines and feature varied mechanisms of action (MOA). They also are more prone to heterogeneity, and subtle differences may occur across product lots, resulting from differences in conditions used in their production processes, including variables such as differences in cellular post-translational modification, cell passage and culture, production and purification.
Good manufacturing practices (GMP) quality control (QC) in vitro potency assays are required for the release of biotherapeutics, applied to demonstrate product performance consistency across lots and time. Bioassays often apply primary or clonal cells cultured under conditions that support modeling of the drug product’s cellular MOA, and often employ complex methods with multiple factors (product, cells, etc.) that influence variability and require highly trained scientists and analysts. To address this, guidance documents support the generation and validation of robust methods based upon a principle of relative potency (USP <111>, Chapters 1032, 1033 and 1034). Assay and test sample acceptance criterion are strictly set to identify good and bad assays and products, relative to method validation limits and characterized biotherapeutic reference standards.
Cells used are cultured via standard techniques, and bioassays are frequently performed without antibiotics because of the potential to affect product-specific responses. Microbial contamination also would alter the cell responses elicited, so successful validation and performance of these bioassays require they be performed by highly trained personnel in laboratories designed, equipped and controlled to prevent culture contamination. Integral to this process is having an effective contamination control (CC) program with analysts trained in the application of aseptic technique (Fresney, Basic Principles of Cell Culture; Culture of Cells for Tissue Engineering, Ch. 1 eds., John Wiley & Sons, Hoboken, NJ, 2006; and Phelan, Basic Techniques in Mammalian Cell Tissue Culture,Curr. Protoc. Cell Biol. 1.1.1 - 1.1.18, September 2007).
GMP QC cell-based bioassay laboratories (QC bio assay labs) do not perform aseptic manufacturing, but, for the reasons described above and because there are no regulations specific to their function, ISO 14644 (class 8), U.S. Food and Drug Administration (FDA) Sterile Drug Products Guidance (2004) and USP <1116>(controlled support), and Eudralex Volume 4 Annex 1 (grade D) manufacturing cleanroom recommendations/requirements are applied. The directives include appropriate design, engineering and process control infrastructure, validated CC and environmental monitoring (EM) programs to ensure both viable (Table 1) and non-viable particulate contamination limits are met, but are intended for facilities that support aseptic manufacturing processes. They greatly exceed the principles generally applied to cell and tissue culture.
Table 1. Regulation or Recommendations for Microbiological "In Operation" cfu Limits
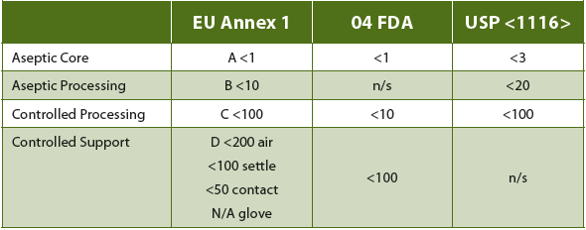

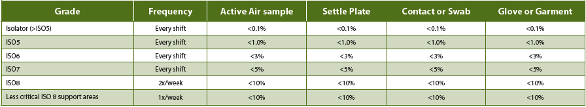
Eudralex Vol. 4 An. 1 (15) directs that grade C and D monitoring “in operation should be based upon the principles of quality risk management.” The “requirements and action/alert limits will depend on the nature of the operations carried out.” While still directed at aseptic manufacturing and support operations, the only defined requirement (18) is that “monitoring should be frequent.” While some flexibility is generally expected and allowed in the recommendations/requirements for measures applied to such classified facilities, only current USP <1116> (38-NF33, 2015) cleanroom guidance suggests that non-sterile applications (such as QC bioassay labs) require different microbial control strategies. In addition, the current USP <1116> has recommended that, rather than defined microbial limits, sampling should be directed toward determination of contamination rates (Table 2) as a better estimation of CC program effectiveness. This is based upon acknowledgement that even ideal cleanroom design and operations cannot prevent all contamination, the source of which is invariably human operators. Expectations for establishing and maintaining a “sterile” environment are recognized in that document to be "technically not possible and unrealistic.” As these, too, are directed toward aseptic manufacturing and processing facilities, this can and does leave application of CC and EM method requirements subject to the interpretation of individual laboratories and regulatory agencies.
Table 2. USP <1116> Recommended Contamination EM Frequency and Incidence Rate Limits
PPD® Laboratories performs GMP QC bioassay testing in three GMPcompliant laboratories in the U.S. and Ireland. All are designed, built, controlled and monitored to comply with the aseptic manufacturing cleanroom controlled support requirements, and extending aseptic cell culture practices beyond those generally recommended (Table 3). PPD’s first QC bioassay lab (B5-156) was commissioned for GMP testing in 2008 at the site in Middleton, Wisconsin, U.S. It consists of a single, 750 ft2 controlled access lab with an exterior gowning/degowning room and operational workflow designed to support the validated CC program. Every biosafety cabinet (BSC) testing event currently includes non-viable particulate and viable settle plate EM. This is supplemented by limited weekly cleaning and EM sampling in BSCs and general lab areas; along with complete monthly cleaning, with full post-cleaning EM in BSCs and pre-and post EM for general lab areas. EM for mycoplasma contamination is performed quarterly. Results from EM are reported semi-annually, and, since implementation, no contamination trends nor any mycoplasma have been detected. There have been only four documented culture contamination events (none impacting QC testing, and none affecting cell bank generation) since recordkeeping was initiated in March 2012, while none have occurred since April 2013.
Table 3. PPD applied CC practices, compared with those described for basic aseptic cell culture principles (italics denote measures not described in basic principles)

In May 2013, PPD commissioned a second 6,200 ft2 controlled access lab suite (B8) in Middleton (GMP certification provided July 2015). A third lab, at PPD’s Athlone, Ireland, GMP facility (Athlone lab) was commissioned in October 2014, and added another 2,000 ft2 of bioassay testing labs to support EU GMP biotherapeutic testing (GMP certification provided January 2015). The design and materials of both suites were based upon B5-156, with controlled workflow and identical CC programs. However, because of the demonstrated effectiveness of the B5-156 program, EM was reduced to complete quarterly pre- and post-cleaning EM in the culture laboratory, and annual pre-and postcleaning EM in the rest of the suite (settle plates were added to the quarterly Athlone bioassay lab EM).
For the B8 site, EM reports are prepared annually, and no significant EM trends or mycoplasma have been detected since initiation of operations. In that laboratory suite, 16 culture contamination events were recorded since commissioning, and increased in frequency from July to September 2014. These did not correlate with an EM viable trending increase, and none impacted the generation of cell banks. In one case, culture contamination resulted in a three-day delay in testing a QC stability sample. As for the B5-156 laboratory, all culture contaminants were identified for genus and species, and all were associated with aquatic environments (all various species of Brevundimonas, Burkholderia, Methylobacterium and Ralstonia).
An investigation into the observed increase in culture contamination events for causes determined the following: 1) the water baths used to warm media and reagents were the likely contamination source; 2) during this period the required cleaning and EM were not being performed per SOPs; 3) increased staffing had doubled lab activities; and 4) cell culture/aseptic technique training of new employees varied by trainer. (Note: Half of the events were associated with training.)
Corrective action responses included: 1) changing the water bath microbicide; 2) retraining of cleaning/EM personnel and boosting EM oversight to ensure compliance; and 3) standardizing cell culture/ aseptic technique training. Since implementation in October 2014, there have been no instances of culture contamination in the laboratory (last event recorded was September 23, 2014).
To demonstrate that the Athlone lab’s application of the U.S. PPD CC and EM programs provided “adequate assurance of control in the environment” compared to other PPD labs performing the same function, a temporary (six month) increase in the EM program (above that applied in the B5-156 lab, with each analyst also performing weekly in-use viable air, surface, equipment and glove EM) was implemented. Prior to the conclusion of that study, the PPD Middleton B8 QC culture laboratory increased gowning (to apply disposable, whole body lab suits and boot covers); performed validation of disinfectant effectiveness and added sporocide to BSC material decontamination; and increased EM to a monthly culture lab frequency (quarterly for support areas) and weekly in-use viable monitoring (as applied temporarily in the Athlone lab). These activities have required a substantial increase in support staffing and have increased analyst time required to perform cell culture by approximately 20%.
Results generated from the Athlone lab six-month increased EM evaluation period (using the original CC program copied from the U.S. sites) and from the first three months of increased B8 culture lab BSC EM confirmed a lack of laboratory and in-use BSC sterility. At both sites, and across 32 analysts and almost 500 cell culture EM events, there was viable contamination observed (Tables 4 and 5), but no mycoplasma was detected. Viable contaminant levels were observed, but no coincidental excursions from PPD’s action/alert limits occurred and only three analyst glove contaminations occurred consecutively across EM events. Over this period, no EM trends were observed, and, in all but four instances (<1 5="" 90="" 93="" all="" and="" aseptic="" athlone="" b8="" b="" bscs="" but="" cfu="" class="" cleanliness="" cleanroom="" colony="" contamination="" controlled="" d="" em-detected="" em="" established="" eu="" events="" facilities.="" fda="" floors="" for="" forming="" grade="" in="" levels="" limits="" m3="" manufacturing="" met="" microbiological="" of="" operation="" outside="" p="" particulate="" processing="" respectively="" source="" support="" targets.="" the="" units="" were="" within="">
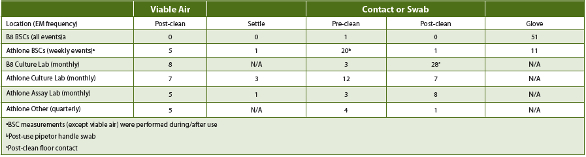
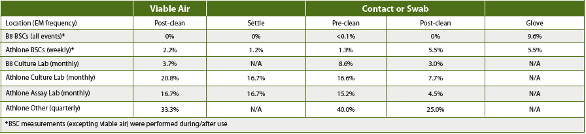

Table 4. PPD B8 Oct-Dec 2015 (Culture Lab only) and Athlone Jan-Jun 2015 Laboratory Contamination Load (high cfu/site)
Table 5. PPD B8 Oct-Dec 2015 (Culture Lab only) and Athlone Jan-Jun 2015 and Laboratory Percent Contamination Frequency
Assessing the increased facility CC and/or EM results based upon USP <1116> contamination frequency guidelines demonstrated that the QC labs’ BSCs met controlled support manufacturing cleanroom contamination frequency limits across the respective periods (Table 5), while the lab areas in Athlone did not. The EM results in Athlone and B8 demonstrated that cleaning procedures reduced, but did not eliminate, contamination events.
During the first three months after the increased BSC CC and EM were implemented, B8 EM observations did show reduced BSC contamination incidence and frequency compared to the Athlone EM during the sixmonth period that the original CC program was applied. However, the B8 settle plate results were similar to historical data collected in the B5-156 cell lab. There also was no indication that the extra CC measures had any impact on glove contamination, when comparing the Athlone and B8 EM results. In addition, the B8 culture lab EM, applying an unchanged CC program outside the BSCs, had ~50% of the observed EM contamination frequency as the Athlone lab.
Despite these EM results and since measures were implemented in the U.S. cell labs after introduction of corrective active measures following the September 2014 internal investigation, zero cell culture contamination events have been recorded at any PPD GMP QC bioassay laboratory.
Conclusions
GMP QC bioassay labs do notperform aseptic manufacturing/processing. However, efficient generation of cell cultures and banks to support such testing does require that effective CC (that incudes analyst training in aseptic technique) and EM programs be implemented to establish and maintain a sufficiently clean environment to support these activities. Another valuable indicator of CC program effectiveness is recording culture contamination events, as these can help establish frequency and identify patterns that might be indicators of a lack of appropriate CC.
CC program effectiveness also can be monitored by a bioassay’s failure rate. The complex and variable nature of these methods requires establishing validated system suitability specifications so as to enable detection of good and bad assays, as well as product. Contamination of cells can and does alter activity and result in assay failure. Repeated assay failures - triggering GMP quality investigations - can support identification of CC system failures.
It is estimated that at least 6,500 GMP QC sample tests have been performed and at least 100 cell banks generated since the initiation of culture contamination records (March 2012). While it is possible that cell culture contamination has resulted in assay failures, none have ever been recorded as a demonstrated cause within PPD’s QC bioassay labs. During this period, PPD’s QC bioassay labs have recorded 20 culture contamination events, all with organism identification related to aquatic environments. As noted previously, half were linked to a failure to follow cleaning and EM procedures over a three-month period and half occurred during analyst training, while none have been recorded since corrections were implemented in October 2014.
Throughout the service history of the PPD QC bioassay labs, application of validated CC measures (that includes recording of culture contamination events and monitoring of assay performance) and EM programs has demonstrated program efficacy equivalence to aseptic manufacturing controlled support cleanroom expectations, with no significant EM trending across sites and time. While culture contamination events have been recorded, only one has had an impact on GMP QC bioassay sample testing (resulting in a stability sample testing window being missed by three days) over a multi-year period.
The results of added EM testing at two sites over a three- and sixmonth period fail to link BSC in-use contamination with culture contamination events, supporting B8 observations in which routine EM did not trend with increased culture contamination events. Further, increased CC measures for that site did not decrease observed glove contamination event frequency, relative to the original CC program, when comparing the EM between the two sites. The increased CC had no impact on culture contamination, but, regardless of the CC program applied, no culture contamination was observed in either laboratory.
It is possible to conclude that, with PPD’s original CC program and its highly trained staff, a reduced EM plan (that includes reporting and assessment of culture contamination and linked assay failure events) is adequate to ensure proper cleaning/disinfection is reducing contamination to a level that allows for effective GMP QC bioassay testing.
In summary, it is appropriate that, in the absence of specific regulations, QC bioassay labs follow available aseptic manufacturingcontrolled support regulations and appropriate application of quality risk management supported by facility performance histories, as directed by Eudralex Vol. 4 An. 1 (15). However, as our results clearly demonstrate, GMP QC bioassay lab CC and EM programs can be effective without meeting the requirements of aseptic manufacturing core or processing cleanrooms, as their expectations for generation and maintenance of a nearly aseptic environment are required toprevent product contamination, while GMP QC bioassay labs need only to keep cell cultures free of microbial contaminants. PPD’s data suggest that an effective CC program that combines EM, monitoring of culture contamination frequency and organism identification, and GMP required fit-for-purpose application and monitoring of rigid and specific test method system suitability are adequate measures for such laboratories. We encourage the initiation of dialogue between industry and regulatory agencies to determine and recommend best practices, so as to reduce the current variability of CC and EM applied and expected programs.
2 comments:
I always like informative blogs and this is one of them. Thanks for sharing and please keep us up to date like this.pcd pharma company
thanks regards
Post a Comment