INTRODUCTION
A shipment of a packaged pharmaceutical product was contaminated during shipment by an accompanying shipment of naphthalene. A method was required to quickly and accurately quantify the levels of naphthalene contamination to determine the usability of the entire pharmaceutical shipment. The Short Path Thermal Desorption System was used in conjunction with the Solid Sampler Oven from Scientific Instrument Services to develop a method to rapidly identify and quantify the naphthalene directly from the solid pharmaceutical product and packaging material. This technique permitted the accurate quantification of the naphthalene in the solid pharmaceutical, without using chemical or other solvent type extraction methodology. Therefore, no impurities from the solvents would be encountered. There wasn't any concern about the efficiency of a solvent extraction technique or loss of material due to vaporization; the disposal problem of used solvents was eliminated.
EQUIPMENT
The samples were collected on the S.I.S. Solid Sampler Oven using Tenax®-TA Traps. For the desorption process, the S.I.S. Short Path Thermal Desorption System Model TD-1 was attached to the injection port of a Varian 3400 Gas Chromatograph. The Gas Chromatograph was interfaced to a Finnigan MAT 8230 high resolution double focusing magnetic sector mass spectrometer. The data were acquired and processed using a Finnigan MAT SS300 data system. The mass spectrometer was operated in the electron ionization mode (70 eV), with a scan of masses 35-350 once each second.
The GC was equipped with a 60 meter x 0.32 I.D. by 0.25 micron film thickness DB-1 capillary column. The GC injection port was maintained at 225 degrees C and a 10:1 split ratio was employed. The GC oven was maintained at -20 degrees C during the sampling and desorption process, after which the oven was temperature programmed from -20 degrees to 280 degrees C at a rate of 10 degrees per minute.
The Tenax TA traps were prepared by packing the Glass Lined Thermal Desorption Tubes with 100 mg of Tenax TA between two silanized glass wool plugs. The packed desorption tubes were then conditioned in the S.I.S. Thermal Desorption Conditioning Oven with a temperature program of 4 degrees per minute from room temperature to 300 degrees C and then held at this upper temperature of 300 degrees for four hours while continually purging with high purity nitrogen at a flow of 10 ml/min. The tubes were then removed from the Conditioning Oven, cooled and sealed with stainless steel caps with PTFE seals until ready for sampling, as described below.
The Thermal Desorption Sample Collection Oven was maintained at an oven temperature of 80 degrees C during sampling with a sparge gas flow of 40 ml/min through the sample. Glass Sample tubes, 1/4 in. x 14 in. long, were used for holding samples with the appropriate fittings for the sparge gas inlet and Desorption Tube attachments (Figure 1).
The Thermal Desorption heater blocks were maintained at 220 degrees C with a carrier gas flow of 10 ml/min through the sample during sample desorption into the GC.
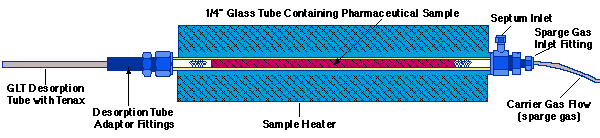
Figure 1 - Cross Section of the Thermal Desorption Sample Collection Oven For the Collection of Thermally Sparged Volatile Samples
EXPERIMENTAL
The pharmaceutical sample consisted of a powdered solid preparation inside gelatin capsules packaged in a plastic bottle with a cotton plug and plastic bottle cap. In addition, bundles of the pharmaceutical bottles were shrink-wrapped for shipment. Due to this method of packaging, the outer bottles were expected to have higher levels of contamination due to their closer proximity to the source of contamination and openings in the shrink wrap film. Bottles from the outside of the bundle are referred to as outer samples, whereas the bottles analyzed from the interior of the bundle were referred to as the inner samples.
The inner and outer containers were opened and the following samples were set up for analysis:
A) The Cotton Plug (Inner & Outer Bottles)
B) Capsules From the Top of the Bottle Just Under the Cotton Plug (Inner Container)
C) Capsules From the Bottom of the Bottle (Inner Container)
The above sampling methodology would permit the determination of the degree of migration of the naphthalene between the inner and outer bottles in the packaging, as well as the migration from top to bottom within the sealed bottles.
The contents of two capsules with 351.5 mg each on average of the pharmaceutical solid matrix were emptied into the 1/4 in. O.D. x 14 in. long glass tube of the S.I.S. Thermal Desorption Sample Collection Oven between two silanized glass wool plugs (Figure 1). Therefore, the total amount of sample used was 702 mg for each analysis. The two samples of cotton analyzed were 12.5 mg for the outer container and 96.8 mg for the inner container. Once the samples were placed in the glass sampling tube, they were spiked with 100 ng of D-8 naphthalene internal standard by syringe injection of 1.0 ul of a 100 ng/ul of a D-8 naphthalene stock solution in methanol through a septum inlet built into the sparge gas inlet fitting of the Sample Collection Oven (Figure 1). The glass tube containing the sample was placed into the Sample Collection Oven. The sparge gas line was attached on one end; a preconditioned Tenax TA adsorbent trap was attached to the opposite end of the glass sample tube using the appropriate fittings supplied with the Thermal Desorption Sample Collection Oven (Figure 1). The samples were heated to 80 degrees C for 30 minutes with a sparge gas flow rate of 40 ml/min of high purity Nitrogen in order to trap the volatile components on the adsorbent Tenax TA trap. The temperature of 80 degrees C was chosen, because the naphthalene would be completely purged from the pharmaceutical sample at this temperature. Also, the pharmaceutical sample itself would not be purged from the sample matrix at this temperature. The total volume of sparge gas of 1200 ml was sufficient to totally purge the naphthalene from the heated sample, but the breakthrough volume for naphthalene on the Tenax trap was not exceeded.
The charged desorption traps were then attached to the Short Path Thermal Desorption System, and a syringe needle was attached. The samples were injected into the GC injection port and thermally desorbed in the GC injection port at desorption block temperature of 220 degrees C for 5 minutes at a purge flow of 10 ml/min and a GC injection split ratio of 10:1.
QUANTIFICATION OF NAPHTHALENE
Quantification of Naphthalene in the contaminated pharmaceutical sample and cotton plugs were accomplished using D-8 naphthalene as a matrix-spiked surrogate internal standard with GC-MS detection. This is by far the most accurate and precise method, as well as the most sensitive method of Quantification. Analytical standards of D-8 naphthalene (Aldrich Chemical Company, Inc.) were prepared from the standard stock solutions of the D-8 naphthalene by diluting this solution with methanol using volumetric glassware to prepare 250 ml of a stock solution of D-8 naphthalene at a concentration of 100 ng/ul. A 10.0 mg sample of the solid naphthalene (Aldrich Chemical Company, Inc.) was accurately weighed and transferred to a 10 ml volumetric flask. The volume of the volumetric flask was adjusted to exactly 10.0 ml using the 100 ng/ul D-8 Naphthalene stock solution. Starting from this solution mixture (1000 ng/ul naphthalene and 100 ng/ul of D-8 naphthalene), a series of log and half log dilutions were prepared down to a final concentration of 1.0 ng/ul of naphthalene with the D-8 naphthalene stock solution as the diluent.
Thus, the following stock solutions were obtained:
| Stock Solution | Naphthalene Conc. | D-8 Naphthalene Conc. |
| Spiking Solution | 0 | 100 ng/ul |
| A | 1000 ng/ul | 100 ng/ul |
| B | 500 ng/ul | 100 ng/ul |
| C | 100 ng/ul | 100 ng/ul |
| D | 50 ng/ul | 100 ng/ul |
| E | 10 ng/ul | 100 ng/ul |
| F | 5 ng/ul | 100 ng/ul |
| G | 1 ng/ul | 100 ng/ul |
One ul each of these solutions were then injected onto the top of the Tenax-TA desorption trap, purged with high purity nitrogen for 30 minutes at 10.0 ml/min, thermally desorbed into the GC and analyzed by the same methodology and conditions as used for the pharmaceutical samples. Mass chromatograms for the molecular ion species of D-8 naphthalene (m/z 136) and naphthalene (m/z 128) were then generated; the resulting data was integrated via the computer software.
A series of analysis blanks containing only D-8 naphthalene internal standard were run after each of the analytical standards and periodically between analyses of the pharmaceutical samples. These blanks were run to assure that no instrument contamination occurred due to sample overloading or instrument contamination. These blanks consisted of Tenax - TA adsorbent tubes run through the entire process of sparging through the solid sampler, thermal desorption, and GC/MS analysis in order to verify the validity of the entire sampling methodology.
RESULTS AND DISCUSSION
The experimental data showed that the stock solutions A and B had driven the mass spectrometer to saturation. Therefore, only the data generated in samples C through G were used in creating the calibration curves. The peak area integrations were used to generate the calibration curves for naphthalene relative to D-8 naphthalene internal standard. The curve was constructed by plotting the ratio of naphthalene peak area / the area of D-8 naphthalene area (y-axis) versus the concentration of naphthalene (x-axis) (Figure 2). A linear calibration curve was obtained with a correlation coefficient of 0.994. Regression analysis of this calibration curve yield the following line equation:
y = 0.0122x + 0.016
Where y = Area of Naphthalene / Area of D-8 Naphthalene
x = Naphthalene Concentration In Ng
Therefore, solving for x yields the following equation:
x = (y - 0.016) / 0.0122
The standard error estimates for y and x variables are 0.0453 and 0.000537, respectively.
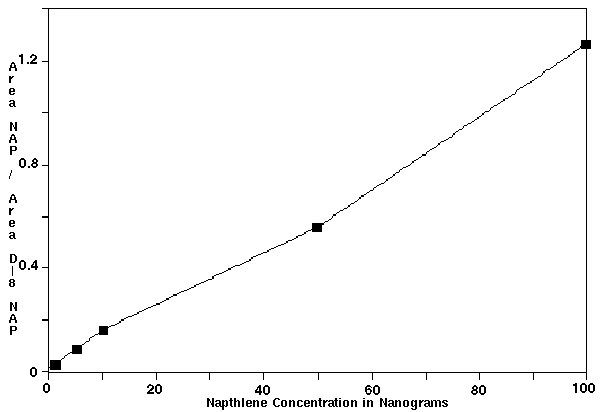
Figure 2 - Calibration curve for the Quantification of Naphthalene
To quantify the naphthalene levels in the pharmaceutical samples and the cotton samples, all the samples were spiked with 100 ng of the D-8 naphthalene, as previously described. The ratio of naphthalene / D-8 naphthalene internal standard peak areas was then determined. The naphthalene concentration was then calculated using the line equation above which was generated from the calibration curve. The concentration of naphthalene in nanograms was then converted to parts per million (ppm) w/w by dividing the values obtained by the individual sample weights in milligrams. The dynamic range of the calibration curve was sufficient to encompass the level of naphthalene encountered in all the samples tested. Based on the data obtained, it was determined that the limit of confirmation for naphthalene is 100 picrograms using this technique. For the 702 milligram pharmaceutical sample this corresponds to less than 1 part per billion (ppb).
PHARMACEUTICAL
A typical total ion chromatograms for one of the pharmaceutical samples is shown in Figure 3c. The single ion mass chromatograms for the m/z 128 (naphthalene) and m/z 136 (D-8 naphthalene) is shown in Figures 3a & 3b for the same sample. When analyzed by the technique described, the GC retention time for D-8 naphthalene is 17.30 minutes and 17.33 minutes for naphthalene. In all the samples analyzed, the relative retention time of naphthalene and its corresponding mass spectrum was found to be identical to that obtained from the analytical standards. Naphthalene was confirmed to be present in all the samples analyzed and was clearly detected at levels much higher than the detection level threshold of 100 picrograms determined above. Blanks, as described previously, contained only the spiked D-8 naphthalene and peaks identified as dimethylpolysiloxane oligomers which arise from the GC septum bleed and column bleed.
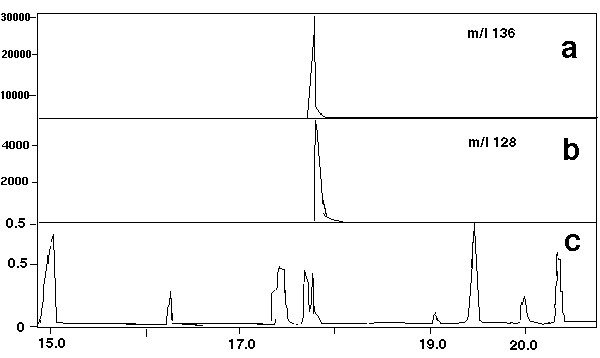
Figure 3 - Single Ion Chromatograms for Naphthalene (m/z 128) and D-8 Naphthalene (m/z 128) and D-8 Naphthalene (m/z 136)
The levels of naphthalene detected in the various pharmaceutical samples and cotton plugs are summarized in Figure 4. The outer containers in the shrink wrapped bundles definitely had higher levels of naphthalene contamination as compared to the inner containers as expected. This was apparently due to the method of packaging which protected the inner packages from contamination by the naphthalene vapors. In all cases, the naphthalene vapors permeated the plastic bottle containers to contaminate the cotton plugs and the pharmaceuticals stored within.
Figure 4 - Table of Results of Quantification of Naphthalene in samples analyzed.
Naphthalene Concentrations in Pharmaceutical Sample Packages
No. Sample Description Naphthalene (ppm)
A-1 Cotton Plug from Outer Container 1.28
A-1 Cotton Plug from Inner Container 0.63
B-1 Pharmaceutical from Outer Container, first bottle 2.52
B-2 Pharmaceutical from Outer Container, second bottle 3.28
C-1 Pharmaceutical from Inner Container, just under cotton plug,
first sample 0.03
C-2 Pharmaceutical from Inner Container, just under cotton plug,
second sample 0.02
D-1 Pharmaceutical from Inner Container, bottom of bottle,
first sample 0.02
D-2 Pharmaceutical from Inner Container, bottom of bottle,
second sample 0.05
The data also indicates that there is no significant difference in the level of naphthalene contamination with respect to the position of the capsules in the container. Capsules from the top of the bottle, just under the cotton plug, contained the same level naphthalene of contamination as those from the bottom of the container.
CONCLUSION
The Short Path Thermal Desorption System used in conjunction with the Solid Sampler Oven permits the identification and accurate Quantification of trace levels of naphthalene in contaminated pharmaceutical products at levels ranging down to 1 ppb with an accuracy of 5.0%. In addition to being very accurate, the technique is rapid, highly efficient, and no chemical or solvent extraction is required. The volatile naphthalene is thermally extracted from the solid pharmaceutical. No contamination from impure solvents contribute to the chromatograms, and sample loss due to solvent extraction efficiency or vaporization of the sample is eliminated. This technique has also been applied to other applications, as the Quantification of benzene and toluene in food products and flavors and fragrances in food products, commercial products, and plants.
No comments:
Post a Comment